A common characteristic of disorders and diseases of the central nervous system (CNS), such as traumatic brain injury (TBI), spinal cord injury (SCI), stroke, Alzheimer's disease, and Parkinson's disease, is the disconnection of axonal pathways and neuronal cell loss1,2,3,4,5,6. For instance, when an ischemic stroke goes untreated, it is estimated that axons are lost at a rate of 7 miles of axons per minute5. In the case of TBI, which approximately 1.7 million people experience each year in the U.S. alone, axonal degeneration may continue to occur years after trauma, as the initial injury precipitates a long-term neurodegenerative state4. Aggravating these deleterious effects, the CNS has a severely limited capacity for regeneration1,7,8,9. Following injury, an inhibitory environment develops that is characterized by a lack of directed guidance to distant targets, the presence of myelin-associated inhibitors that hinder neurite outgrowth, and the formation of a glial scar by reactive astrocytes8,10,11,12. The glial scar serves as a biochemical and physical barrier to regeneration, with molecules such as chondroitin sulfate proteoglycans obstructing axon outgrowth8,11. Furthermore, even though neural stem cells have been found in the adult CNS, the production of new neurons is limited, as consistent evidence of neurogenesis has only been found in the olfactory bulb, the hippocampal subgranular zone, the periventricular area, and the central canal of the spinal cord13,14. These obstacles prevent the functional recovery of lost neurons and white matter architecture following injury or disease, resulting in the often life-changing and prolonged effects of these conditions.
Despite the lack of regenerative capacity in the adult CNS, it has been demonstrated that axonal regeneration is possible if adequate environmental cues are presented to host neurons15,16,17,18. Researchers have attempted to deliver and manipulate growth factors (e.g., nerve growth factor, epidermal growth factor, glial-dependent growth factor, and neurotrophic factor-3) and other guidance molecules to stimulate plasticity and axon regeneration14,18,19. Even though these studies have confirmed that adult axons are capable of responding to growth factors, these strategies are limited by the low permeability of the blood-brain barrier and the specific spatial and temporal gradients required to promote regeneration14,18,19. Other approaches have relied on the hyperactivation of regeneration-related transcription factors in CNS neurons. For example, overexpression of the Stat3 transcription factor stimulated axonal regeneration in the optic nerve20. Nevertheless, both biomolecule delivery and overexpression of transcription factors fail to replace lost neuronal populations. Cell-based strategies have mainly centered on transplanting neural stem cells (NSCs) into the CNS, taking advantage of their capacity to replace CNS neurons, release trophic factors, and support the attempts at neurogenesis that occur after injury17. In spite of this, there are still pressing challenges hindering this approach, including the hampered ability of transplanted neural cells to survive, integrate with the host, and remain spatially restricted to the injured area6,14,17,21. In addition, cell delivery alone is incapable of reinstating the cytoarchitecture of damaged or lost axonal pathways. An alternative approach that addresses the problems facing cell and drug/chemical delivery strategies is combining these approaches with the use of biomaterials14,22,23. Biomaterials such as hydrogels are capable of emulating the biochemical and physical properties of the extracellular matrix (ECM), aiding in cell delivery and retention within the injured area, and delivering growth factors and other bioactive molecules with controlled release22. The attractive characteristics of these biomaterial-based strategies have resulted in evidence of in vivo axonal regeneration after the transplantation of scaffolds to the lesioned area24,25,26,27,28,29,30. However, acellular biomaterial strategies do not replace lost neuronal populations; when used as delivery vehicles for neuronal, glial, or neuronal precursor cells, biomaterials are incapable of reconstituting long-distance axonal networks. The challenge of developing an approach that tackles both the axonal pathway degeneration and neuronal loss associated with CNS injury and disease still remains31.
Our research group previously reported the development of implantable micro-tissue engineered neural networks (micro-TENNs), which are a type of "living scaffold" consisting of neuronal cell bodies restricted to one or both ends of an agarose hydrogel-ECM micro-column, with aligned axonal tracts extending throughout the interior of this three-dimensional (3D) encasement1,10,31,32. One of the main differences between this technique and previous approaches is that the cytoarchitecture of micro-TENNs is created completely in vitro and is transplanted afterwards33,34,35,36,37,38,39,40,41. In vitro fabrication offers extensive spatial and temporal control of cellular phenotype and orientation, mechanical/physical properties, biochemical cues, and exogenous factors, which benefits the integration of these scaffolds with the host after implantation41,42. Micro-TENNs are anatomically inspired because they emulate brain neuroanatomy, displaying axonal tracts similar to those that bridge distinct functional regions of the brain (Figure 1A)1. Therefore, this strategy may be able to physically replace lost white matter tracts and neurons following implantation into a lesioned region. This technique is also inspired by developmental mechanisms in which "natural living scaffolds" formed by radial glial cells and pioneering axons act as pathfinding guides for cell migration from the subventricular zone and axonal outgrowth, respectively43. These mechanisms are recapitulated in the aligned axonal tracts of micro-TENNs, which can present living pathways for neural cell migration and axonal regeneration by axon-mediated axonal outgrowth (Figure 1C)43. Furthermore, this strategy takes advantage of synaptic integration between the micro-TENN neurons and native circuitry, forming new relays that may contribute to functional recovery (Figure 1B)43. The capacity for synapse formation may also grant this approach the ability to modulate the CNS and respond to host tissue according to network feedback. For example, optogenetically active neurons in the living scaffolds may be stimulated to modulate host neurons through synaptic interactions (Figure 1D).
In addition, the biomaterial-based tubular construction of micro-TENNs offers an adequate environment for cell adhesion, growth, neurite extension, and signaling, while the miniature dimensions of the constructs potentially permit minimally invasive implantation and provide a partially sequestered microenvironment for gradual integration into the brain. Indeed, recent publications have demonstrated the potential of micro-TENNs to mimic neural pathways following implantation into the rat brain. Following stereotaxic microinjection, we previously reported evidence of micro-TENN neuronal survival, maintenance of axonal tract architecture, and neurite extension into the host cortex out to at least 1 month in vivo10,31. Moreover, labeling with synapsin provided histological evidence of synaptic integration with native tissue10,31. Overall, micro-TENNs may be uniquely suited to reconstruct and modulate damaged CNS by replacing lost neurons, synaptically integrating with host circuitry, restoring lost axonal cytoarchitecture, and, in certain cases, providing regenerating axons with the appropriate pathfinding cues.
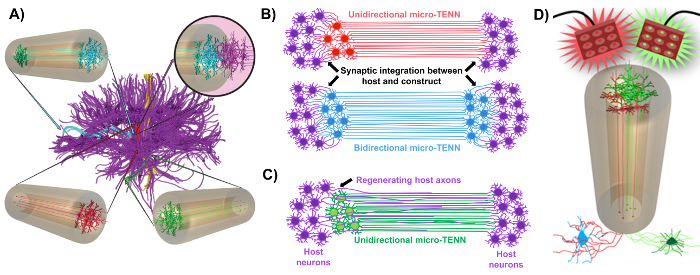
Figure 1: Principles and inspiration behind the development of micro-tissue engineered neural networks (micro-TENNs). (A) Micro-TENNs mimic the cytoarchitecture of the brain connectome (purple), in which functionally distinct regions are connected by long, aligned axonal tracts in a unidirectional (red, green) or bidirectional (blue) manner. As an example, micro-TENNs could reconstitute lost connections in corticothalamic and nigrostriatal pathways or in the perforant pathway from the entorhinal cortex to the hippocampus (adapted from Struzyna et al., 2015)1. (B) Diagram of a unidirectional and bidirectional micro-TENN (red and blue, respectively) synaptically integrating with the host circuitry (purple) to serve as a functional relay between both ends of a lesion. (C) Schematic of the axonal tracts of a unidirectional micro-TENN (green) serving as a guide for axon-facilitated regeneration of host axons (purple) towards a target with which the micro-TENN interacts. (D) Conceptual diagram of the use of optogenetically active micro-TENNS as neuromodulators, taking advantage of synaptic integration with excitatory or inhibitory neurons (bottom). Please click here to view a larger version of this figure.
The current manuscript details the methodology utilized to fabricate micro-TENNs using embryonically derived cerebral cortical neurons. Notably, micro-TENNs could be fabricated with other types of neural cells. For example, the initial reports of successful micro-TENN development featured dorsal root ganglion (DRG) neurons32. The hydrogel micro-columns can be generated (Figure 2A) by adding liquid agarose to a custom-made, laser-cut cylindrical channel array or to capillary tubes, both containing aligned acupuncture needles. The needle forms the lumen and determines the inner diameter (ID) of the micro-column, while the capillary tube ID and the diameter of the cylinders in the laser-cut device dictate the outer diameter (OD) of the constructs. The OD and ID can be chosen according to the desired application by selecting different diameters for the device/capillary tubes and the acupuncture needles, respectively. The length of the micro-columns can also be varied; to date, we have reported the construction of micro-TENNs up to 20 mm in length10 and are actively pursuing even longer lengths. After the agarose gels and the acupuncture needles are removed, an ECM solution generally consisting of type I collagen and laminin is added to the lumen of the constructs (Figure 2C). The ECM core provides a scaffold to support neuronal cell adhesion and axonal outgrowth. Initially, primary rat cortical neurons were plated in the micro-columns using dissociated cell suspensions10,31,32. However, this approach did not produce the target cytoarchitecture in all cases, which was defined as the neuronal cell bodies restricted to the ends of the micro-columns, with the central lumen comprised of pure aligned axonal tracts. Since then, the use of a forced neuronal aggregation method (based on protocols adapted from Ungrin et al.) has enabled a more reliable and consistent fabrication of micro-TENNs with the ideal structure (Figure 2B)44. In addition to describing the current methodology, this article will show representative phase-contrast and confocal images of micro-TENNs that demonstrate the formation of axonal tracts over time, as well as the finalized target cytoarchitecture. This manuscript will also expand on noteworthy aspects of the protocol and remaining challenges and future directions of the micro-TENN technology.
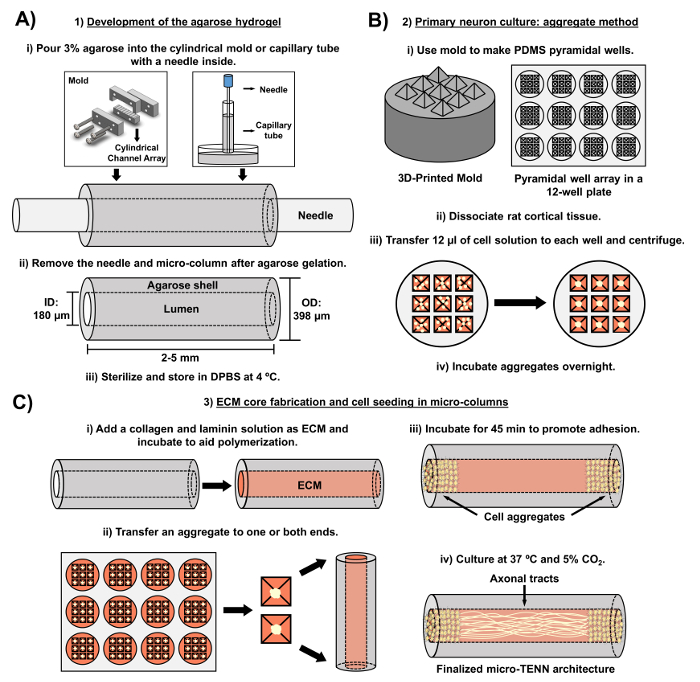
Figure 2: Schematic diagram of the three-stage micro-TENN fabrication process. (A) Development of the agarose hydrogel: (i) Initially, a small acupuncture needle (e.g., 180-350 µm in diameter) is inserted into the cylindrical channels of a custom-made, laser-cut mold or a capillary tube (e.g., 380-700 µm in diameter). In the next step, liquid agarose in DPBS is introduced into the cylindrical channels or capillary tubes. (ii) After the agarose gels, the needle is removed and the mold is disassembled to yield the hollow agarose micro-columns. (iii) These constructs are then sterilized and stored in DPBS. (B) Primary neuron culture and the aggregate method: (i) Neuronal aggregation is performed in pyramidal micro-well arrays, cast from 3D-printed molds, that fit in the wells of a 12-well culture plate. (ii) Micro-TENNs include primary rat neurons dissociated from fetal brains of embryonic-day-18 rats. Following tissue dissociation with trypsin-EDTA and DNase I, a cell solution with a density of 1.0-2.0 x 106 cell/mL is prepared. (iii) 12 µL of this solution are transferred to each well in the pyramidal micro-well array. The plate containing these micro-wells is centrifuged to produce cell aggregates. (iv) These are then incubated overnight prior to plating in the micro-columns. (C) ECM core fabrication and cell seeding: (i) Prior to cell seeding, an ECM solution containing 1 mg/mL type I collagen and 1 mg/mL laminin is transferred to the interior of the micro-TENNs and allowed to polymerize. (ii) Depending upon whether unidirectional or bidirectional micro-TENNs are being fabricated, an aggregate is placed at either one or both extremes of the micro-column, respectively. (iii) Following a period of incubation to promote adhesion, micro-TENNs are cultured in Petri dishes flooded with supplemented embryonic neuronal basal medium. (iv) After 3-5 days in culture, the final micro-TENN structure should demonstrate cell aggregates at the extremes of the micro-column, with axonal tracts spanning its length. Please click here to view a larger version of this figure.
Micro-TENNs were monitored using phase-contrast microscopy to assess their cytoarchitecture and axonal outgrowth (Figure 4). Within unidirectional, 2 mm long micro-TENNs, neuronal aggregates were restricted to one end of the micro-column and projected a bundle of axons through the inner core. Axons spanned the entire length of the column by 5 days in vitro (DIV) (Figure 4A). There was a greater initial axonal growth rate in bidirectional 2 mm-long micro-TENNs, as axons spanned the entire micro-column by 3 DIV (Figure 4B). By 5 DIV, bidirectional micro-TENNs featured dense axonal tracts that connected the aggregates maintained at the extremes of the micro-column. This cytoarchitecture was also observed in 5-mm micro-TENNs, with axons spanning the micro-column by 5 DIV (Figure 4C). As observed in all instances, the ECM in the lumen and the geometric restriction presented by the agarose provided the directionality required to form axonal tracts that structurally mimic features of native neuroanatomy.
Immunocytochemistry techniques were applied in this protocol, and confocal images were taken to verify the components of the micro-TENN architecture. Cell nuclei stained with Hoechst (blue) were localized almost exclusively within aggregates at the extremes of unidirectional and bidirectional micro-columns, with essentially no Hoechst staining in the interior (Figure 5). This observation confirmed what was inferred from the phase-contrast images: neuronal populations were restricted to the ends, and only axons (in red) spanned the lumen of the micro-TENNs. However, after axons crossed the aggregates, cell migration was observed along the axonal tracts into the inner lumen, as observed by the presence of nuclei (blue) in the interior at 28 DIV for a 2 mm-long bidirectional micro-TENN (Figure 6). Furthermore, synapsin I immunolabeling (green) was found throughout the cell bodies and axonal tracts (Figure 6). Synapsin I is a pre-synaptic terminal protein implicated in the regulation of neurotransmitter release and is indicative of the presence of pre-synaptic terminals when found in a punctate distribution; it has been previously correlated with the formation of active synapses by whole-cell patch recordings49. The synapsin staining in the axon-rich central zone of the micro-TENN is likely a combination of puncta as well as synapsin being transported down axons towards pre-synaptic terminals. Nevertheless, the presence of synapsin puncta within the micro-TENN aggregates suggests that these constructs have the capacity to communicate through synapses, although the colocalization of synapsin with a post-synaptic protein would be required for further structural evidence of functional synapses.
Micro-TENNs were also fabricated with aggregated neurons that were transduced with an AAV containing the GCaMP6f calcium indicator to preliminarily probe the electrophysiological properties of these constructs. These constructs expressed the calcium indicator throughout their entire length, as shown by fluorescence in both the neuronal aggregates and the axonal tracts (Figure 7A). Note that due to microscope settings aimed at maximizing the intensity in the aggregates, the fluorescence in the axonal tracts appeared fainter. These transduced micro-TENNs were analyzed over time with high-speed fluorescence microscopy (without external electrical stimulation), and several cell-sized regions of interest (ROIs) were selected at random in a representative micro-TENN to characterize the signaling capacity of these constructs. Calcium concentration bursts were observed in almost all ROIs, as reflected with associated fluctuating fluorescence intensities throughout time (Figure 7B). Particularly, various ROIs seemed to show a near-constant periodicity of calcium concentration increases (Figure 7B). These calcium concentration changes are inferred to be the result of spontaneous, inherent action potentials, cell signaling within individual aggregates, and/or signaling across axons from distinct aggregates. Further analyses are required to fully characterize the electrophysiological properties of neurons and axonal tracts within micro-TENNs. Nevertheless, these initial results demonstrate that micro-TENNs exhibit robust endogenous/baseline electrical activity.
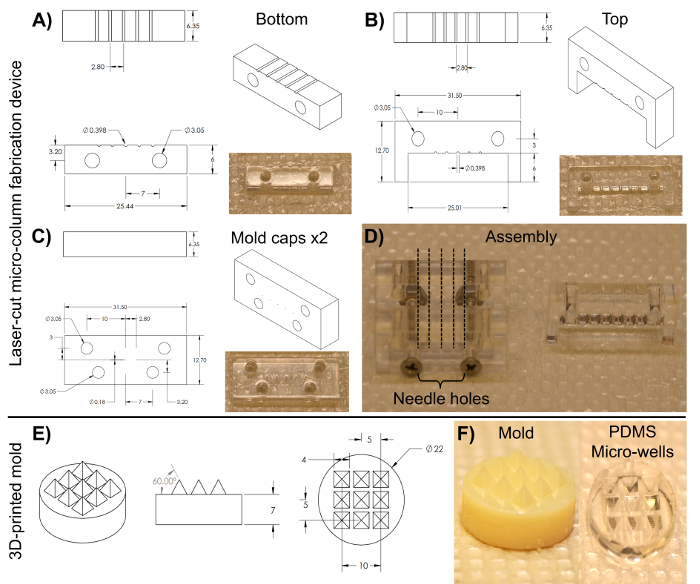
Figure 3: Blueprints of the laser-cut micro-column fabrication device and the 3D-printed pyramidal micro-well mold for cell aggregation. (A)-(B) Blueprint and image of the bottom and top halves of the cylindrical channel array, respectively, used to fabricate the hydrogel micro-columns. (C) Blueprint and image of the caps used to hold the device together. Both anterior and posterior pieces share the same dimensions. (D) Image of the assembled device required to fabricate the micro-columns. Only the two caps and the bottom half are clamped together with screws in the lower two alignment holes (left). The broken lines show the placement of the acupuncture needles through the five holes on each cap. The liquid agarose is added to the bottom half of the cylinders and, afterwards, the top half (right) is placed on the device to mold the liquid agarose into shape. (E) Blueprint of the 3D-printed molds used to make PDMS micro-well arrays. (F) Images of the 3D-printed mold (left) and the PDMS micro-well arrays (right). All dimensions are in millimeters (mm). Please click here to view a larger version of this figure.

Figure 4: Observation of axonal growth in micro-TENNS by phase-contrast imaging of unidirectional and bidirectional constructs as a function of days in vitro. (A) Unidirectional 2 mm long micro-TENNs show axonal tracts extending almost the entire length of the micro-column by 5 DIV. (B) Compared to unidirectional micro-TENNs, bidirectional 2 mm micro-TENNs display a faster axonal growth rate. (C) For 5 mm bidirectional micro-TENNs, axonal tracts populate the length of the micro-column at 5 DIV. This architecture is maintained at least until 10 DIV. Scale bars = 200 µm. Please click here to view a larger version of this figure.
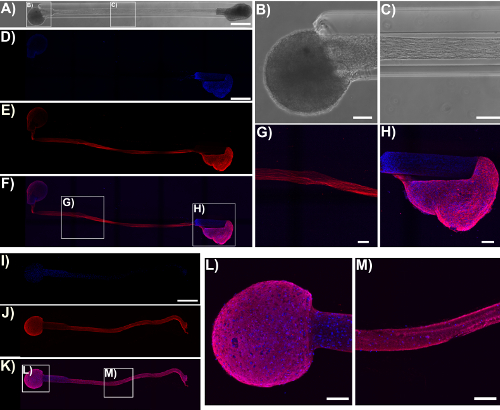
Figure 5: Micro-TENN cytoarchitecture observed with confocal images of immunolabeled unidirectional and bidirectional micro-TENNs. Cells were stained for cell nuclei (Hoechst; blue) and axons (Tuj1; red). (A) Phase-contrast image of a 5 mm bidirectional micro-TENN (28 DIV). Confocal reconstructions (D)-(F) and (I)-(K) of a 5 mm bidirectional (28 DIV) and a unidirectional (25 DIV) micro-TENN, respectively, show labeled cell nuclei (D), (I) and axons (E), (J), as well as the overlay (F), (K). Scale bars: 500 µm. Insets in (A), (F), and (K) refer to phase-contrast (B)-(C) and confocal (G)-(H), (L)-(M) zoom-ins of neuronal aggregates and axonal tracts. The images show the aggregates restricted to the ends of the micro-TENN, while the lumen is spanned by aligned axonal tracts. Scale bars = 100 µm. Please click here to view a larger version of this figure.
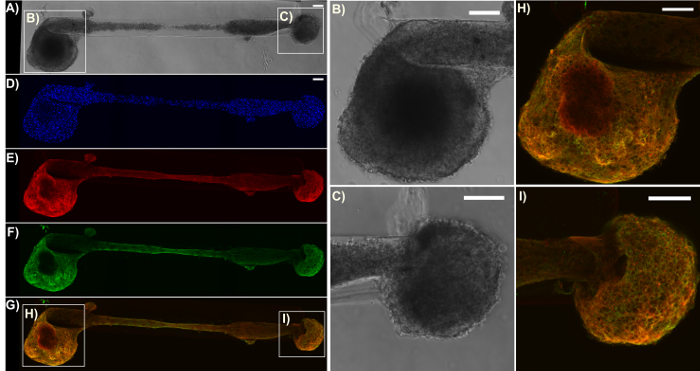
Figure 6: Expression of the pre-synaptic terminal protein synapsin I in a representative bidirectional micro-TENN. The construct was stained for cell nuclei (Hoechst; blue), axons (Tuj1; red), and pre-synaptic boutons (synapsin I; green). (A) Phase-contrast image of a 2 mm bidirectional micro-TENN at 28 DIV. (D)-(G) Confocal reconstructions showing the cell nuclei (D), axons (E), pre-synaptic boutons (F), and an overlay (G) of the three channels. Boxes show zoom-ins (B)-(C), (H)-(I) of the phase-contrast and overlay images for the neuronal aggregates, respectively. Scale bars = 100 µm. Please click here to view a larger version of this figure.
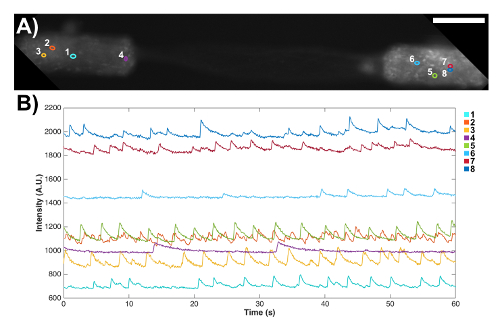
Figure 7: Spontaneous signaling activity in a bidirectional micro-TENN observed through the expression of a genetically encoded calcium indicator. (A) Fluorescence microscopy confirmed the expression of the GCaMP reporter, as observed by fluorescence throughout the aggregates and axons. Scale bar = 100 µm. (B) Fluorescence changes associated with calcium concentration variations were measured without external stimulation at several regions of interest (ROIs) as a function of time. The numbered colored circles in (A) designate these ROIs. Please click here to view a larger version of this figure.

Figure 8: Phase-contrast images of common modes of failure in the micro-TENN methodology. (A) A completely dehydrated/dried agarose micro-column as a result of the removal and/or evaporation of DPBS in the initial stages of fabrication. Scale bar = 50 µm. (B) Hydrogel micro-column with the lumen lining the outer wall of the construct due to the lack of concentric alignment of the acupuncture needle with the capillary tube. Scale bar = 100 µm. (C) Seeded micro-TENN with both aggregates present at 1 DIV. (D) One of the aggregates fell off from the same micro-column as (C) at 3 DIV. (E) Unidirectional micro-TENN at 4 DIV. (F) The aggregate and axonal tracts fell out from the micro-column in (E) and remained floating in the culture medium at 5 DIV. Scale bar for (C)-(E) = 300 µm. Please click here to view a larger version of this figure.
