To gain insights into the formation of F-actin patches on early endosome membranes, we followed the protocol outlined in Figure 2. Briefly, cells were transfected with GFP-RAB5 and then early endosomes were prepared by subcellular fractionation. These purified early endosomes were incubated with cytosol in order to provide actin itself as well as other factors possibly involved in the reaction. At the end of the incubation period, the reaction was stopped by fixation of the mixture. A sample was then collected and deposited on a microscopic slide. Finally, F-actin was revealed with phalloidin (Figure 3A). The nucleation of F-actin onto early endosomal membranes as well as the subsequent polymerization process occurred rapidly. Indeed, F-actin could already be visualized on endosomes within 1min after the beginning of the reaction (Figure 3A). These actin structures, which were initially short, rapidly became longer, branched and linked to one another leading to the formation of an interwoven network of actin filaments (Figure 3A), presumably reflecting unbalanced actin dynamics in vitro. Similar nucleation and polymerization rates were observed when the assay was carried out with purified rhodamine-labeled actin in addition to cell cytosol. This was done in glass-bottomed dishes so that structures could be imaged directly, in the absence of any perturbation (Figure 3B). In the assay described here, early endosomes nucleate de novo actin polymerization selectively. Indeed, actin polymerization did not occur when the in vitro reaction was carried out in the absence of either endosomes or cytosol. It can thus be concluded that early endosomes possess the fundamental ability to support the nucleation and subsequent polymerization of actin filaments9,11.
To analyze the amount of actin polymerized from endosomes, images acquired at different times were analyzed using CellProfiler (Figure 4A). The amount of actin polymerized per endosome was measured as the area of actin surrounding a single early endosome. The actin polymerization rate was higher at the beginning of the process and decreased with time (Figure 4B). The actin network could be analyzed in more detail at early time-points after the beginning of the reaction. At these early time-points, individual actin-containing structures could be still be differentiated from each other. In order to evaluate the complex organization of actin networks, the number of actin filaments emanating from each endosome was counted (i.e., primary filaments). Similarly, the number of actin filaments originating from primary filaments (i.e., secondary filaments) were counted, as well as the number of all other actin filaments that could be identified in the networks and that did not originate from endosomes (i.e., other filaments) (Figure 4C-E. Once the actin filaments were traced, other parameters, such as length of individual actin filaments and number of endosomes connected to the same actin filaments, could be easily calculated (Figure 4F).
Figure 1: Phase Contrast Micrograph of a HeLa Cell Homogenate. After homogenization, the extract was mounted between a microscopic slide and a coverslip. For publication, micrographs were captured with a 100X objective after oil immersion. For routine inspection, however, the cell extract was visualized under a 20X or a 40X objective without oil immersion. Two examples of high-magnification views of the homogenate are shown (A-B). Nuclei (star) appear as round structures dark gray in color, without attached cell remnants. Arrows point to the characteristic granules of varying size, shape, and translucence, which are released upon cell homogenization and correspond to intracellular materials (i.e., organelles). Scale bar = 10 µm. Please click here to view a larger version of this figure.
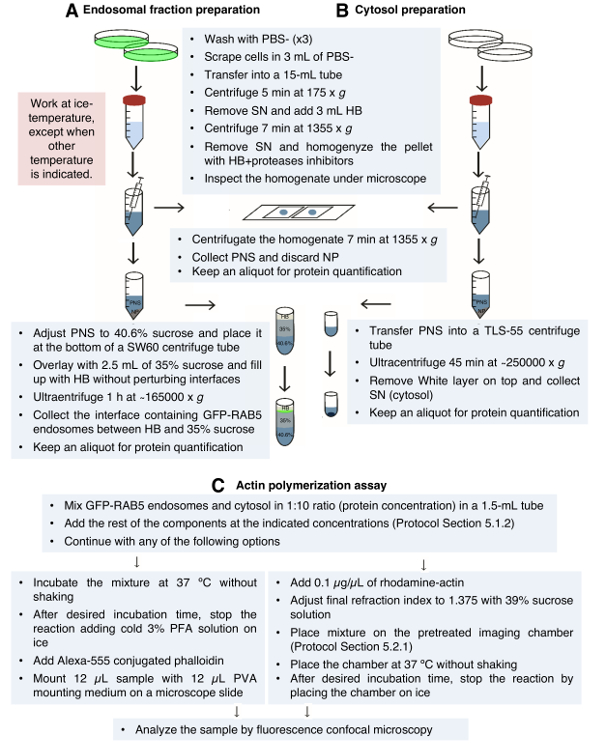
Figure 2: Schematic Representation of the Protocol. Schematic description of the protocols used to prepare the endosomal extracts (A) and cytosol extracts (B) and to monitor actin polymerization from endosomes in vitro (C). Supernatant (SN), homogenization buffer (HB), postnuclear supernatant (PNS), nuclear pellet (NP), and paraformaldehyde (PFA). Please click here to view a larger version of this figure.
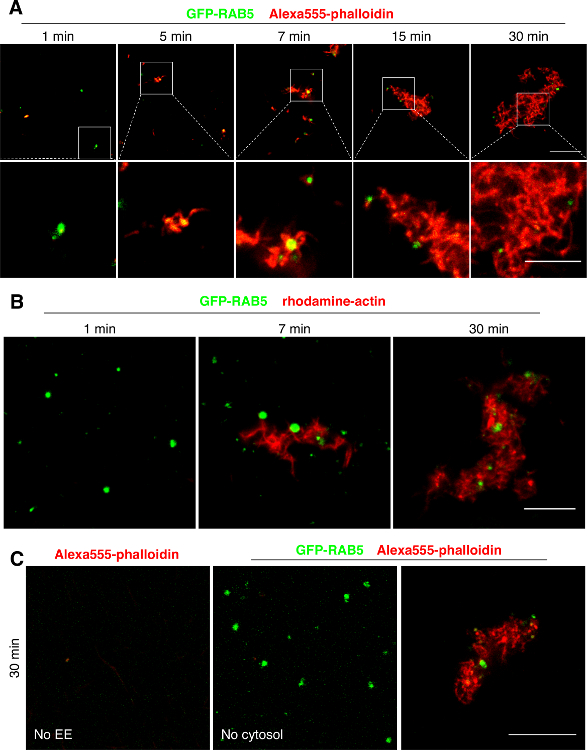
Figure 3: Nucleation and Polymerization of F-actin on Early Endosomes In Vitro. (A) Endosomes purified from Hela cells expressing GFP-Rab5 (green) and HeLa cytosol were prepared separately. In the assay, early endosomes (EEs) were mixed with cytosol, and the mixture was incubated for the indicated times. The mixture was then fixed, labeled with phalloidin (red) to label F-actin, and analyzed by fluorescence confocal microscopy.Bars: 10 µm (top panel) and 5 µm (bottom panel). (B) EEs purified from Hela cells expressing GFP-Rab5 (green) and HeLa cytosol were prepared separately. These cell extracts were incubated with purified rhodamine-actin (red) for the indicated times in microscope chambers and were analyzed by confocal microscopy. Scale bar = 10 µm. (C) The experiment was performed as in A. HeLa cytosol (without EEs) and HeLa endosomes (green) labeled with GFP-Rab5 (without cytosol) were incubated separately (left and middle panels) or together (right panel) for 30 min, fixed, labeled with phalloidin (red) to label F-actin, and analyzed by fluorescence confocal microscopy. Scale bar = 10 µm. Please click here to view a larger version of this figure.
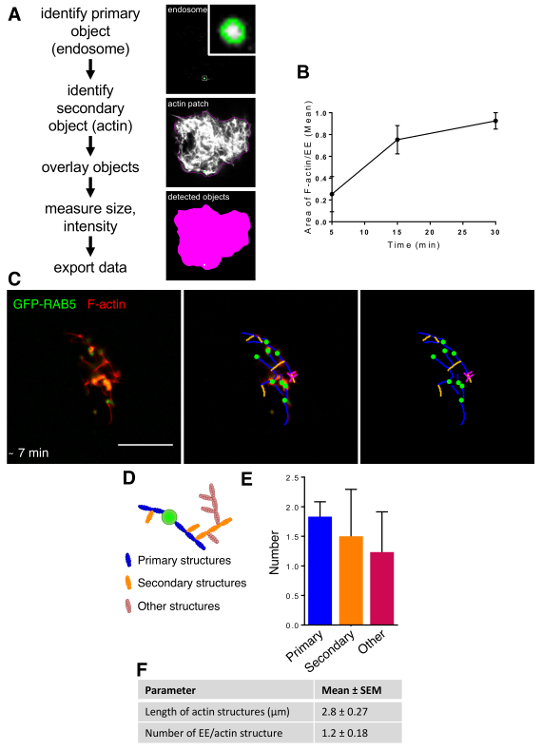
Figure 4: Analysis of the Actin Network. (A) Workflow of CellProfiler analysis. (B) Number of polymerized actin were quantified relative to the number of endosomes using the CellProfiler software. Data are median ± s.e.m. (n= 3). Data are means ± s.d. (n = 3). 5 min versus 15 min (ns P= 0.0736), 5 min versus 30 min (* P = 0.0195), and 15 min versus 30 min (ns P = 0.3129). (C) Early endosomes (EEs) purified from Hela cells expressing GFP-Rab5 and HeLa cytosol were prepared separately, and the assay was carried out as in Figure 2A. Samples were fixed after 7 min. The middle panel shows the same micrograph, with the overlap of the traces of F-actin structures drawn using the NeuronJ plugin13 of the ImageJ software. The right panel shows the traces only. Scale bar = 10 µm. (D)Representation of the F-actin structure numbering sequence used in the quantification with ImageJ software. (E) Quantification of the number of actin filaments, as defined in C-D. Data are means ± s.d. (n = 3). Primary branches versus secondary branches (ns P = 0.5262), secondary branches versus other branches (ns P = 0.6815), and primary branches versus other branches (ns P = 0.2254). (F) The length of F-actin structures and the number of EEs per actin structure are shown as examples of other parameters than can be analyzed with the quantification defined in C-D. Please click here to view a larger version of this figure.
