Representative data for the thermolabile ligand DSC are shown in Figure 1. The position and height of the thermolabile ligand-bound peak successively shifts down towards that of the unbound biomolecule as the thermolabile ligand is depleted with each scan (Figure 1a). The free denaturation profile is used as a reference for the endpoint of thermolabile ligand conversion (Figure 1b). Data for MN4 bound to quinine are shown as a negative control for thermolabile ligand conversion (Figure 1b). The final thermolabile ligand scans have slightly higher transition midpoints and peak heights relative to the unbound MN4, indicating the thermolabile ligand conversion product (benzoylecgonine) has a weak affinity for MN4. The thermodynamic parameters resulting from global analysis of the datasets in Figure 1 are listed in Table 1. The folding parameters for MN4 in the presence of cocaine or quinine are identical within error, as expected since dilute small molecules are not expected to perturb the folding thermodynamics of the free biomolecule. The binding parameters are in good agreement with those from isothermal titration calorimetry (ITC)18 and reveal that MN4's preference for quinine over cocaine is driven by a more favorable binding enthalpy. In Figure 2, increasing the high temperature equilibration period yields more pronounced reductions of the thermolabile ligand concentration with each scan relative to the short equilibration period dataset. Using the optimized global fit concentration parameters from the two datasets, the rate constant for ligand conversion at the high equilibration temperature is calculated from the slope of the line in the Figure 2 inset.
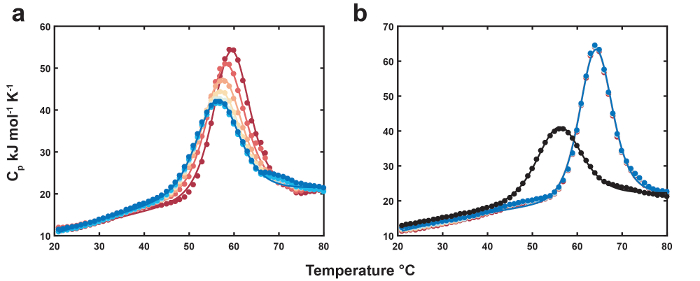
Figure 1. Thermolabile ligand DSC. (a) Thermograms of MN4 (83 µM) bound to cocaine (initial concentration 778 µM). First and last scans of MN4 in the presence of ligand are shown as dark red and blue circles while the corresponding fits are shown as colored lines. (b) Thermograms of free MN4 (83 µM, black circles) and successive scans bound to quinine (880 µM, colored circles). Fits to the free and quinine-bound datasets are shown as black and colored lines, respectively. Reproduced from reference4 with permission from the Royal Society of Chemistry. Please click here to view a larger version of this figure.
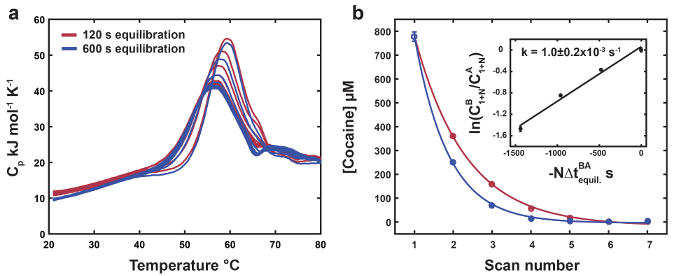
Figure 2. Measuring the Rate Constant for Thermolabile Ligand Conversion. (a) Sets of thermograms for MN4 bound to cocaine with short (120 s, dark red) and long (600 s, dark blue) equilibration times at 80 °C, respectively. (b) Concentrations of cocaine extracted from global analysis of the datasets in (a) as a function of scan number. Experimental points and exponential fits are shown as colored circles and lines respectively. The inset shows a linear fit to the previously described Supplementary Eq. 19 from Harkness et al. using the optimized global fit cocaine concentrations for the two datasets4. The rate constant for ligand thermal conversion at 80 °C is calculated as the slope of the line. The error for the rate constant for thermolabile ligand conversion is given as ± two standard deviations. Reproduced from reference4 with permission from the Royal Society of Chemistry. Please click here to view a larger version of this figure.
| Fit parameters | Cocaine added | Quinine added |
| ∆HUF | 271.3±1.8 | 272.5±4.0 |
| ∆SUF | 824.4±5.1 | 827.9±10.9 |
| a∆GUF | 21.6±0.2 | 21.6±0.9 |
| a∆HB1F | -75.2±1.6 | -101.0±4.0 |
| a∆SB1F | -154.2±5.0 | -213.7±12.0 |
| ∆CpB1F | -1.5±0.1 | -1.2±0.1 |
| a∆GB1F | -28.5±0.2 | -36.2±0.7 |
| a∆HB2F | -33.7±1.8 | – |
| a∆SB2F | -49.9±5.2 | – |
| ∆CpB2F | -2.2±0.1 | – |
| a∆GB2F | -18.6±0.3 | – |
Table 1. Thermodynamic Parameters Extracted from Global Analysis of the MN4 DSC Datasets Using Cocaine and Quinine Ligands. Parameters were calculated at 30 °C. B1F refers to cocaine- or quinine-bound folded states and B2F refers to the benzoylecgonine-bound folded state. ΔH and ΔG are expressed in kJ/mol, ΔS is expressed in J/mol/K and ΔCp is expressed in kJ/mol/K. Errors were calculated according to the variance/co-variance method19.
Modifications and troubleshooting
The details of the global fitting analysis used in Figure 1 and Figure 2 have been described previously4. Here, we outline practical aspects of performing and analyzing DSC binding experiments with thermolabile ligands. Note that a DSC baseline obtained for the thermolabile ligand alone is subtracted from the ligand + biomolecule dataset, effectively cancelling out the heat released or absorbed by the thermal conversion process itself. The standard thermolabile ligand global fitting analysis (Figure 1 and Figure 2) assumes that the system is at thermodynamic equilibrium throughout the temperature scan and that the thermolabile ligand concentration is constant throughout each thermogram, decreasing exclusively during the high temperature equilibration period. We have previously shown that this assumption applies to cocaine-bound MN4 and is expected to hold for any thermolabile ligand/biomolecule system with kinetics similar to these.
There are, however, some situations in which the system cannot be assumed to be at thermodynamic equilibrium and/or the concentration of ligand cannot be considered constant throughout a single scan. These include i) when the ligand thermally converts rapidly relative to the temperature scan rate, ii) when the biomolecule undergoes irreversible aggregation at high temperature, iii) when the folding/unfolding rates are slow compared to the scan rate, and iv) when the ligand association/dissociation rates are slow compared to the scan rate. In these cases, the system is under kinetic rather than thermodynamic control and the analysis given in reference 4 cannot strictly be applied. Data may be simulated quantitatively following Figure 3, as described in the Supplementary File 1. In principle, these kinetics-based calculations could be used to fit non-equilibrium DSC data, potentially yielding both kinetic and thermodynamic data, however this analysis is beyond the scope of this paper. Instead, we present some representative simulated DSC data to assist the reader in identifying non-equilibrium situations.
An ideal example of thermodynamic control is shown in Figure 4a, b. DSC thermograms of the free biomolecule are superimposable (Figure 4a) and scans with the thermolabile ligand do not show hysteresis, such that the melting temperature observed on the up-scan matches the folding temperature of the previous down-scan (Figure 4b). When the thermolabile ligand converts rapidly compared to the scan rate, large distortions appear in the thermogram and the thermodynamic equations do not account for the peak shape, as shown in Figure 4c, d. This can be alleviated somewhat by increasing the scan rate. When the biomolecule aggregates in a temperature-dependent manner, DSC traces for the free biomolecule show successive decreases in magnitude (Figure 4e), while addition of the thermolabile ligand produces a pattern of decreasing thermal upshifts similar to the ideal case, but scaled by the decreasing biomolecule concentration (Figure 4f). When folding/unfolding kinetics are slow compared to the scan rate, hysteresis is apparent in DSC traces of the free biomolecule such that the apparent denaturation temperature on the up-scan is higher than the apparent renaturation temperature on the down-scan (Figure 4g). Addition of a thermolabile ligand leads to the familiar pattern of decreasing thermal upshifts, particularly for the up-scans (Figure 4h). Finally, systems with rapid folding and slow binding produce hysteresis-free DSC thermograms for the free biomolecule (Figure 4i), however data with the thermolabile ligand show hysteresis where the apparent melting temperature of the up-scan is higher than the apparent folding temperature of the previous down-scan (Figure 4j). Nevertheless, the typical pattern of decreasing thermal upshifts are apparent in both up-scans and down-scans. Non-equilibrium behavior in the case of slow folding or binding kinetics can be alleviated somewhat by decreasing the scan rate, although this runs the risk of non-negligible ligand thermal conversion occurring throughout the scan. In practice, the scan rate and upper equilibration temperature can be adjusted manually to obtain data resembling Figure 4a, b.
Limitations of the technique
Our thermodynamic analysis for DSC binding experiments with thermolabile ligands requires that the folding and binding processes are relatively rapid and that thermolabile ligand conversion is slow prior to the high temperature portion of each scan. When the lifetime of the folded and/or bound state is greater than about 30 s (koff, ku < 0.03 s-1), hysteresis becomes discernable in scans performed at 1 °C min-1. Additionally, when the ligand conversion rate constant is above approximately kc = 10-4 s-1 before the denaturation transition, there can be significant depletion of the ligand during the course of a single scan. Application of our analysis is also inappropriate when irreversible aggregation occurs. In these cases, more advanced modelling could be applied to the data. No affinity information is available if ligand conversion is so rapid that it reaches completion prior to the first denaturation transition.
Significance with respect to existing methods
Our method for the first time allows DSC to be used to measure the binding thermodynamics of high affinity, thermolabile ligands. By performing a global simultaneous analysis of all scans, thermodynamic parameters are extracted with high accuracy20. An additional benefit is that the full dataset can be collected in as little as one experiment if the thermal conversion product has no affinity for the free biomolecule. In contrast, producing a typical experimental DSC series for a non-thermolabile ligand requires ~ 7-10 total experiments.
Future applications
This approach has direct applications to characterizing tight, thermolabile inhibitors in drug discovery campaigns. Several therapeutic compounds such as antibiotics and benzodiazepines are known to be thermolabile, undergoing rapid hydrolysis at or near physiological pH and temperatures of ~ 60-70 °C11. This DSC method is well positioned to identify and characterize many more. As well, modification of the fitting protocol to account for systems under kinetic rather than thermodynamic control, as discussed above, has the potential to open the door to many more systems of biological relevance.
Critical steps within the protocol
One of the most important experimental procedures to consider is dialysis or exchange of the biomolecule and ligand into identical working buffer solutions (protocol steps 1.3-1.6). Buffer mismatch between the ligand and biomolecule solutions can lead to large artifacts in the baseline and sample scans which completely obscure the relevant folding data. Additionally, it is essential that the power reading stabilizes before the DSC is pressurized so that it can be monitored during pressurization (protocol step 2.3). If the power reading changes by more than ~ 10 µW during pressurization, bubbles have likely formed in the capillaries and can cause large artifacts in the data. In this case, the solutions need to be degassed more thoroughly.
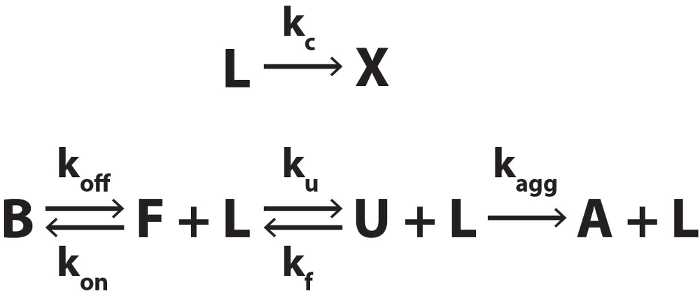
Figure 3. Biomolecular Folding, Binding to a Thermolabile Ligand, and Irreversible Aggregation. The thermolabile ligand (L) converts to product (X) with a rate constant kc. X has no affinity for the biomolecule. The bound state (B) of the biomolecule exchanges with the free folded state (F) with rate constants koff and kon. F exchanges with the unfolded state (U) with rate constants ku and kf. U irreversibly converts to the aggregated state (A) with the rate constant kagg. Please click here to view a larger version of this figure.
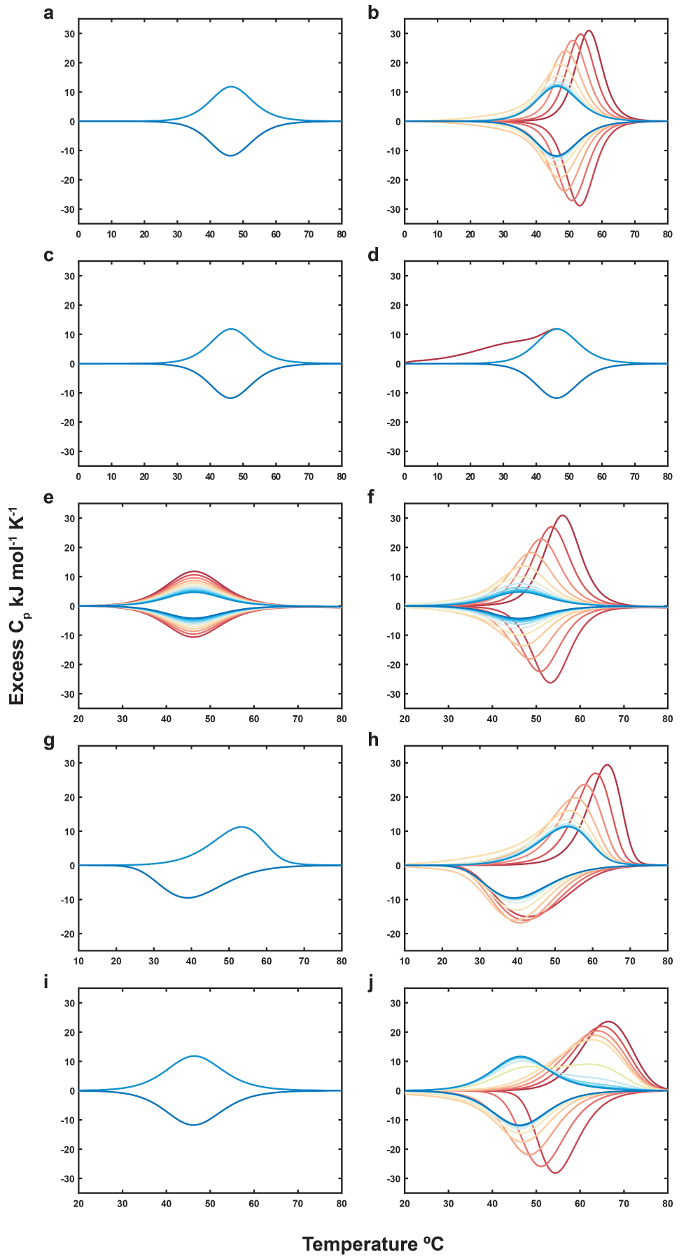
Figure 4. Computer Simulation of Equilibrium and Kinetically-controlled DSC Experiments in the Absence and Presence of a Thermolabile Ligand. (a) Equilibrium biomolecular folding. (b) Equilibrium folding, thermolabile ligand binding, and slow thermolabile ligand conversion. (c) Equilibrium biomolecular folding. (d) Equilibrium folding, thermolabile ligand binding, and fast thermolabile ligand conversion. (e) Equilibrium biomolecular folding and slow irreversible aggregation. (f) Equilibrium folding, binding, slow thermolabile ligand conversion, and slow irreversible aggregation. (g) Slow biomolecular folding. (h) Slow folding, equilibrium binding, and slow thermolabile ligand conversion. (i) Equilibrium biomolecular folding. (j) Equilibrium folding, slow thermolabile ligand binding, and slow thermolabile ligand conversion. In all panels, the first and last simulated scans are dark red and dark blue, respectively. Panels that show only light and dark blue thermograms indicate that all simulated scans overlay, and only the last two are visible in the plot. DSC experiments were simulated with 20 scans (10 melting and 10 annealing) at 1 °C min-1 scan rate, with a temperature range of 0-80 °C. Certain panels display narrower temperature ranges to allow better visualization of the simulation trends. The biomolecule and ligand concentrations in the simulations were 200 µM and 10 mM, respectively. Each experiment was simulated with a 600 s equilibration time at 0 °C before scanning and a 60 s equilibration time between each of the subsequent scans. Arrhenius parameters for equilibrium binding and folding were Aon = 5 x 10-1 M-1 s-1, Aoff = 1 x 1019 s-1, Eaon = -20 kJ mol-1, Eaoff = 120 kJ mol-1, Afold = 1 x 10-14 s-1, Aunfold = 5 x 1018, Eafold = -80 kJ mol-1, and Eaunfold = 120 kJ mol-1. Arrhenius parameters for kinetically-controlled binding and folding were Aon = 5 x 10-3 M-1 s-1, Aoff = 1 x 1016 s-1, Eaon = -20 kJ mol-1, Eaoff = 120 kJ mol-1, Afold = 1 x 10-16 s-1, Aunfold = 5 x 1016, Eafold = -80 kJ mol-1, and Eaunfold = 120 kJ mol-1. Arrhenius parameters for slow and rapid thermolabile ligand conversion were Aslow = 7.509 x 1010 s-1, Easlow = 94.65 kJ mol-1, and Afast = 1 s-1, Eafast = 10 kJ mol-1. Arrhenius parameters for slow irreversible aggregation were Aagg. = 5 x 107 s-1 and Eaagg. = 80 kJ mol-1. Excess heat capacities were calculated with ΔHUF (unfolding), ΔHBF (binding), and ΔHAF (aggregating) = 200, -140, and 50 kJ mol-1, respectively. The theoretical description of kinetically-controlled DSC experiments with thermolabile ligands and script for performing these simulations (and the necessary parameters) are available in Supplementary File 1. Please click here to view a larger version of this figure.
