1. Fluorescence-based Screening of Incorporation Efficiencies (Figure 1)
- Maintain HEK293 cells in Dulbecco's Modified Eagle's Medium (DMEM; high glucose, 4 mM glutamine, pyruvate) supplemented with 10 % (v/v) fetal bovine serum (FBS), 100 U/mL penicillin and 100 µg/mL streptomycin at 37 °C, 95 % humidity and 5 % CO2.
- Seed the cells the day before transfection.
- Detach the cells for 5 min at 37 °C in 0.05 % Trypsin/PBS supplemented with 0.5 mM EDTA. Use 1 mL Trypsin/EDTA for a 10-cm dish. Quench with 10 volumes of complete medium and resuspend the cells by pipetting. Count the number of cells in the suspension using a hemocytometer49.
- Seed 6.0 x 105 HEK293 cells per well of 6-well plates in 2 mL complete growth medium. Prepare as many wells as the number of samples, and two additional wells for the wild-type EGFP and a mock-transfected sample, respectively.
- Control confluence (area occupied by the cells) under a microscope. Transfect cells at ~70 % confluence using polyethyleneimine (PEI) reagent.
- 1h prior to transfection, add the appropriate amount of freshly prepared ncAA stock solution to all wells for a final ncAA concentration of 0.25-0.5 mM. Add the ncAA to all wells, including the wild-type positive control and mock-transfected cells, to prevent differences in fluorescence signals that may be caused by effects of the ncAA on cellular growth.
Note: To prepare stock solutions, dissolve the ncAA to 0.1-0.5 M using 0.2-0.5 M NaOH. However, some ncAAs may require initial solubilization in DMSO and/or neutralization by four volumes of 1 M HEPES (pH 7.4) before use. Commonly, the manufacturer recommends a protocol to prepare a stock solution. - In a microcentrifuge tube, mix 1 µg of plasmid DNA encoding for the ncAARS/tRNA pair to be tested with 1 µg of reporter plasmid DNA (pcDNA3.0-EGFP183TAG-mCherry). In separate tubes, prepare an identical transfection using the EGFP wild-type reference and a mock transfection.
Note: Number of copies of the tRNA cassette embedded in the plasmid encoding for the ncAARS/tRNA pair depends on the application. To facilitate cloning, 1 tRNA copy is recommended when screening different tRNAs, whereas 4 copies are recommended (albeit not strictly necessary) when either testing different ncAARS or the incorporation of different ncAAs by the same orthogonal pair. - To each tube containing the DNA add 100 µL lactate buffered saline (LBS) containing 20 mM sodium lactate at pH 4.0 and 150 mM NaCl. Mix briefly.
- To each tube containing the DNA in LBS add 6 µL of 1 µg/µL PEI in LBS (ratio PEI/DNA = 3/1 w/w) and vortex immediately. Incubate at RT for 10-15 min.
- Take 400 µL cell medium from each well and add it to the DNA-PEI mixture to neutralize the pH. Dribble the DNA mixture onto the cells.
Note: DMEM usually contains phenol red as pH indicator. During the neutralization step the color of the mixture added in the tube will change from yellow (acidic) to red (neutral). Although forming the DNA complexes in LBS at acidic pH gives the highest transfection yields50, DNA-PEI complexes can alternatively be formed directly at pH 7.4 (for instance in serum-free DMEM). If using DMEM to form DNA complexes, skip the neutralization step 1.3.5. In any case, it is essential that no serum is present in the mixture when forming the complexes.
- 1h prior to transfection, add the appropriate amount of freshly prepared ncAA stock solution to all wells for a final ncAA concentration of 0.25-0.5 mM. Add the ncAA to all wells, including the wild-type positive control and mock-transfected cells, to prevent differences in fluorescence signals that may be caused by effects of the ncAA on cellular growth.
- Harvest cells 48 h post-transfection.
- Aspirate the medium and rinse the cells once with 2 mL pre-warmed PBS (37 °C). Add 800 µL of PBS supplemented with 0.5 mM EDTA and incubate for 20 min at 37 °C. Detach and suspend the cells by pipetting up and down.
- Transfer the cell suspension into 1.5 mL tubes containing 200 µL PBS supplemented with 5 mM MgCl2.
- Centrifuge for 2 min at 800 x g and discard the supernatant.
Note: The protocol can be paused here. In this case, flash-freeze the pellets in liquid N2 and store at -80 °C for up to one month. Always wear eye protection goggles.
- Add 100 µL Tris lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA and freshly added PMSF) to the cell pellets and incubate on ice for 30 min. To facilitate lysis, vortex every 5 min.
- Spin down the cell debris for 10 min at 4 °C and 14,000 x g and transfer 90 µL of the supernatant into black 96-well plates. Measure EGFP and mCherry fluorescence using a plate reader equipped with a fluorescence module.
Note: Use appropriate excitation and emission filters for EGFP (λabs: 488 nm; λem: 509 nm) and mCherry (λabs: 588 nm; λem: 611 nm). Measured EGFP values will span a range between the minimum value obtained from mock-transfected cells and a maximum value, which is usually obtained from wild-type EGFP. Take care of setting up the correct measurement window in the instrument. - The efficiency of ncAA incorporation is calculated as the ratio between the fluorescence of the sample and the fluorescence obtained from expression of wild-type EGFP. All values are normalized to mCherry fluorescence.

2. Genetic Incorporation of ncAAs into GPCRs for Photo-crosslinking Mapping of Ligand-GPCR Interactions (Figure 3)
- Maintain HEK293T cells in DMEM supplemented with 10 % (v/v) FBS, 100 U/mL penicillin and 100 µg/mL streptomycin at 37 °C, 95 % humidity and 5 % CO2.
- Seed cells the day before transfection.
- Detach the cells for 5 min at 37 °C in 0.05 % Trypsin/PBS supplemented with 0.5 mM EDTA. Use 1 mL Trypsin/EDTA for a 10-cm dish. Quench with 10 volumes of complete medium and resuspend the cells by pipetting up and down. Count the number of cells in the suspension using a hemocytometer49.
- Seed 5.0 x 105 293T cells per well in 2 mL complete growth medium in 6-well plates. For each position to be screened, prepare 1 well per ligand plus one well for the binding control33,38. An extra well to be transfected with the wild-type (wt) receptor may be included to check the expression level of the mutant.
- The day after, control confluence (area occupied by the cells) under a microscope. Transfect cells at ~70% confluence using PEI.
- 1h prior to transfection, add Azi to all wells to a final concentration of 0.5 mM.
- Prepare a 0.5 M stock solution of Azi. Per 6-well plate, weigh 1.2 mg Azi into a tube and dissolve it in 15 µL 0.5 M NaOH. Dilute the stock solution in 1.2 mL complete medium and add 200 µL of the mixture to each well.
Note: Prepare a fresh stock solution of Azi for every experiment. The azide moiety has a short half-life in aqueous solutions, especially at basic pH, and the AziRS incorporates the intact but also the degraded form.
- Prepare a 0.5 M stock solution of Azi. Per 6-well plate, weigh 1.2 mg Azi into a tube and dissolve it in 15 µL 0.5 M NaOH. Dilute the stock solution in 1.2 mL complete medium and add 200 µL of the mixture to each well.
- Transfect a total amount of 2 µg DNA per well: 1 µg of plasmid encoding for the FLAG-tagged GPCR bearing a TAG codon at the desired position and 1 µg of the plasmid encoding for the orthogonal pair dedicated to Azi (E2AziRS51 and 4 copies of the cognate suppressor-tRNA BstYam)33,38.
Note: When including a wt comparison to check expression levels, transfect a lower amount of plasmid DNA for the wt receptor. Depending on the GPCR, 0.2-0.5 µg of plasmid encoding the wt receptor yield similar levels as 1.0 µg of the mutant plasmid. Transfect the same amount of DNA in all wells, filling up the missing DNA with a mock (for instance an empty vector). - Proceed as described in 1.3.3-1.3.5.
- 48 h post-transfection, proceed either with step 2.4 for photo-crosslinking of the ligands or go to step 2.5 for direct harvesting and analysis for verifying receptor expression.
- 1h prior to transfection, add Azi to all wells to a final concentration of 0.5 mM.
- Photo-crosslinking of the ligand.
- Prepare a 1,000x ligand stock solution. Dissolve the peptide ligand at a concentration of 100 µM in DMSO.
Note: The ligand concentration depends on the dissociation constant KD of the ligand-GPCR interaction. A final concentration of 100 x KD is recommendable. If the peptide ligand is a salt of trifluoroacetic acid (TFA), consider the weight of TFA when calculating the molecular weight (1 x TFA per basic amino acid in the peptide). Also, consider that peptides are in general hygroscopic. Avoid repeated freezing of peptide powder and never open a peptide container until it has not reached room temperature. - Dilute the ligand stock solution 1:1,000 in binding buffer consisting of 0.1% BSA, 0.01% Triton-X 100, 5 mM MgCl2 in HEPES dissociation buffer (HDB) containing 12.5 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-HCl pH 7.4, 140 mM NaCl and 5 mM KCl. Prepare 1 mL per Azi-GPCR mutant. Replace the cell medium with 1 mL of the ligand solution. Incubate for 10 min at RT.
Note: Adjust the incubation time to the specific GPCR, accounting for ligand kinetics and receptor internalization. Prolonging the incubation time does not improve crosslinking yields. - Irradiate the samples for 20 min in a UV crosslinker at 365 nm with 5 x 8 W tubes and ~ 5 cm distance to the cells. Detach the cells by pipetting and transfer them into a 1.5 mL reaction tube. Pellet the cells for 3 min at 800 x g and discard the supernatant.
- Dissolve a tablet of protease inhibitor (PI) cocktail in 1 mL 25 mM EDTA/H2O to make a 50x stock solution. Aliquot the PI stock solution and store it at -20 °C. Dilute the 50x stock 1:25 in HDB and resuspend the cell pellets in 50 µL of 2 x PI in HDB. Flash-freeze the cells in liquid N2.
Note: Wear eye protection goggles. At this point, the samples can be stored at -80 °C for up to one month. Proceed with step 2.6.
- Prepare a 1,000x ligand stock solution. Dissolve the peptide ligand at a concentration of 100 µM in DMSO.
- Direct cell harvest.
- Aspirate the medium. Add 800 µL of 0.5 mM EDTA in HDB. Incubate for 10 min at RT or on ice.
- Detach the cells by pipetting up and down and transfer them into a 1.5 mL reaction tube. Add 200 µL of 5 mM MgCl2 in HDB. Pellet the cells for 3 min at 800 x g and discard the supernatant.
- Resuspend the cell pellets in 50 µL of 2 x PI in HDB and flash freeze in liquid N2. Wear eye protection goggles.
Note: At this point, the samples can be stored at -80 °C for up to 1 month.
- Cell lysis.
- Thaw the cells in a water bath at 37 °C for 30-45 s and vortex briefly. Keep samples cold from now on. Pellet membranes at 2,500 x g and 4 °C for 10 min. Discard the supernatant, which contains the bulk of cytosolic proteins.
- Resuspend the pellets in 50 µL HEPES lysis buffer containing 50 mM HEPES-HCl pH 7.5, 150 mM NaCl, 10 % glycerol, 1 % Triton X-100, 1.5 mM MgCl2, 1 mM EGTA, 1 mM DTT and freshly added 2 x PI cocktail. Mix thoroughly. Lyse the cells 30 min on ice and vortex every 5 min.
- Spin down the cell debris for 10 min at 14,000 x g and 4 °C. Immediately transfer the supernatant to a fresh reaction tube.
Note: Proceed with the analysis right away. The lysates can be stored at -20 °C, however, every freeze-thaw cycle impairs the quality of the results.
- Western blot analysis.
- To prepare the sample, take 3-5 µL lysate and fill it up to 7 µL with H2O. Add 2 µL 1 M DTT and 3 µL 4 x sample buffer containing 63 mM Tris-HCl pH 6.8, 2 % SDS, 10 % glycerol and 0.04 % bromphenol blue. Incubate for 30 min at 37 °C.
- When the GPCR is glycosylated and faint or smeared bands are a problem, deglycosylate samples with PNGase F to increase signal intensity and sharpen the bands. Use 3-5 µL lysate and deglycosylate in a total volume of 10 µL following the supplier's protocol. Add 3 µL 4 x sample buffer.
Note: Membrane proteins are often glycosylated in multiple sites and states, which impairs the quality of resolution in SDS-PAGE analysis. However, do not deglycosylate the samples for analysis of the expression level of the Azi-GPCR mutants using anti-FLAG antibodies because it is relevant to evaluate the portion of the fully glycosylated, mature receptor at the cell surface. - Resolve samples via standard SDS-PAGE and blot transfer proteins to a PVDF membrane.
CAUTION: Acrylamide is neurotoxic. Wear gloves and eye protection. - Block the membrane for 1 h at RT or overnight at 4 °C in 5 % skim milk in TBS-T containing 20 mM Tris-HCl pH 7.4, 0.15 M NaCl and 0.1 % Tween 20.
- Probe the membrane with an anti-ligand antibody followed by the HRP-conjugated secondary antibody. Wash in between with TBS-T. To detect the expression level of the Azi-GPCR, probe the membrane with a commercial HRP antibody (see Table of Materials).
- Perform enhanced chemiluminescence (ECL) reaction using homemade ECL reagent and detect signals for 5 min in the dark.
3. Ultrafast Bioorthogonal Labeling of GPCRs on Live Mammalian Cells
Note: The protocol is optimized for 4-well chambered coverslips (well area = 2.2 cm2). For different well sizes, the protocol must be scaled accordingly.
- Surface coating of microscope slides. Carry out the whole procedure under a sterile hood.
- Prepare a poly-D-lysine hydrobromide (MW=500-550 kDa) (PDL) stock solution at a concentration of 1 mg/mL in 50 mM borate buffer (pH 8.5). Store at 4 °C for up to 6 months. Do not freeze.
- Dilute the PDL stock solution 1:40 in sterile ultra-pure water to a final concentration of 25 µg/mL (working solution), then filter the solution through a 0.22 µm sterile filter.
Note: The working solution can be stored at 4 °C for up to 3 months. - Fully cover the bottom of each well of the microscopy slide with 500 µL of PDL working solution. Incubate for 20 min at RT and aspirate the PDL working solution.
Note: The PDL working solution can be used up to three times. If the solution needs to be reused, transfer the used solution from the coated slides to a fresh sterile tube and label the tube accordingly. Never mix the recycled solution with fresh solution. - Rinse each well 3 x with ~ 700 µl sterile ultra-pure water and let dry for at least 1 h.
Note: It is very important to rinse the wells accurately, as residues of the PDL solution are toxic to the cells. The coated slides can be used straight away for microscopy or stored for up to one week at 4 °C.
- Maintain HEK293T cells in DMEM supplemented with 10 % (v/v) FBS, 100 U/mL penicillin and 100 µg/mL streptomycin at 37 °C, 95 % humidity and 5 % CO2.
- Seed cells the day before transfection.
- Detach the cells for 5 min at 37 °C in 0.05 % Trypsin/PBS supplemented with 0.5 mM EDTA. Use 1 mL Trypsin/EDTA for a 10-cm dish. Quench with 10 volumes of complete medium and resuspend the cells by pipetting. Count the number of cells in the suspension using a hemocytometer49.
- Seed 1.0 x 105 HEK293T cells per well (area 2.2 cm²) in 600 µL dye free complete DMEM.
Note: For imaging purposes, it is very convenient to work from the beginning in a medium that does not contain any dye. Dye free DMEM formulations are commercially available.
- Control confluence (area occupied by the cells) under a microscope and transfect the cells at ~70 % confluence using a lipid-based transfection reagent.
- 1 h prior to transfection, prepare a fresh 100 mM stock solution of TCO*K in 0.2 M NaOH and 15 % (v/v) DMSO.
- Per well, mix 3 µl of the TCO*K stock solution with 12 µL of 1 M HEPES pH 7.4. Gently add the solution to the wells for a final TCO*K concentration of 0.5 mM.
- Prepare a total amount of 500 ng DNA per well. In a microcentrifuge tube, dilute 200 ng of pcDNA3.1_CRF1R-95TAG-EGFP, 200 ng of the plasmid encoding for the MbPylRSAF/tRNAPyl orthogonal pair (four cassettes of tRNAM15) and 100 ng of pcDNA3.1_Arrestin3 plasmid in 50 µL medium (dye free, serum-free, antibiotic free).
Note: In general, co-transfection of Arrestin is not necessary to observe GPCR internalization. However, co-transfecting Arrestin3 speeds up internalization of CRF1R, which is very convenient when analyzing internalization of many mutants. - Dilute 1.25 µL of the lipid-based transfection reagent (2.5 µL per 1 µg of DNA) in 50 µL medium (dye free, serum-free, antibiotic free) and add the solution to the DNA mixture. Vortex immediately and incubate 5-10 min at RT. Add DNA-lipid complexes to the cells.
Note: In our experience, the morphology of cells transfected using lipid-based transfection looks more physiologic compared to that of cells transfected with PEI. As PEI gives higher transfection efficiency, PEI should be preferred for downstream applications like Western blot, whereas lipid-based transfection is a better choice to transfect cells for imaging experiments.
- 24 h post-transfection, label the receptor with fluorescent dyes.
- Prepare a 0.5 mM dye-tetrazine stock solution in DMSO and a 10 mg/mL of DNA staining dye stock solution in ultra-pure H2O.
- Transfer 100 µL medium from each well into a 1.5 mL reaction tube. Add 1.8 µL of the dye-tetrazine stock solution and 0.3 µL of the DNA staining dye stock solution. Transfer the medium containing the dyes back to the well and incubate for 5 min at 37 °C.
Note: Tetrazine-orange-fluorescent dye has a final concentration of 1.5 µM. - Aspirate the medium and gently rinse the cells twice with PBS to remove excess of dye. Add 600 µL of complete dye free growth medium preheated to 37 °C.
- Fluorescence microscopy and receptor internalization.
- Visualize the labeled receptors under 63x (or similar) magnification using filters appropriate for GFP (λabs: 488 nm; λem: 509 nm), orange-fluorescent dye (λabs: 550 nm; λem: 570 nm) and DNA staining dye (λabs: 350 nm; λem: 461 nm). Take a picture with each filter before activating the receptor.
- Promote receptor internalization using 200 nM of Ucn1.
- Prepare a 1,000 x Ucn1 stock solution of 200 µM in DMSO.
Note: Depending on the solubility of the peptide, you may be able to prepare the stock in pure water or buffer. - Transfer 100 µL medium from a well into a 1.5 mL reaction tube and add 0.6 µL of the peptide agonist stock solution. Transfer the medium back into the well.
- Observe the internalization under the microscope. Take pictures after the clearly detectable occurrence of internalization (10-15 min to hours, depending on the receptor and overexpression of Arrestins) using the filters mentioned previously.
- Prepare a 1,000 x Ucn1 stock solution of 200 µM in DMSO.
The outline of the fluorescence assay is depicted in Figure 1. The assay is employed in three applications. In first place, a number of tRNA variants for incorporation of Lys(Boc) by the Pyl orthogonal pair are screened. Lys(Boc) is an amino acid sterically similar to Pyl. As Pyl is not commercially available, Lys(Boc) is commonly used as a standard substrate for the PylRS. The screened tRNAs are based on the tRNAPyl. Each tRNA variant bears mutations of single bases or base-pairs in the loops and stems rationally designed to improve tRNA stability and compatibility with the eukaryotic translational machinery. All details about tRNA design are described in our recent work30. Typical results are described in Figure 2A: different tRNAs give different suppression efficiency, which is clearly reflected in the amount of measured fluorescence. The small error bars of biological triplicates show that the values are highly reproducible. In second place, two gene variants of the E2AziRS26,51, which incorporates the photo-crosslinker p-azido-Phe (Azi), are evaluated. E2AziRS is derived from the E. coli TyrRS. One gene variant employed native E. coli codon usage, whereas the second was codon-optimized for the use in H. sapiens. The fluorescence assay shows that the codon-optimized variant gives higher protein yields (Figure 2B). Finally, the incorporation rate of different amino acids for bioorthogonal click chemistry are compared (Figure 2C). All the ncAAs presented here (BCNK, TCO*K and SCOK) are incorporated by the same orthogonal MbPylRSAF/tRNAPyl pair. Lys(Z) is also incorporated by this pair and was used as a positive control. To illustrate the reliability of the fluorescence assay, parallel ncAA incorporation experiments were performed on a GPCR, the CRF1R. Western blot analysis was carried out to estimate GPCR expression (Figure 2B, C). The same trends observed with EGFP in the fluorescence assay were observed with the CRF1R. Notably, Azi incorporation into CRF1R was highly enhanced when using the codon-optimized E2AziRS gene compared to the native gene, showing that even a moderate improvement of 1.5-2-fold for a soluble protein (EGFP) according to the fluorescence assay can have a greater impact on the expression level of more challenging membrane proteins (Figure 2B). As general note, with respect to the number of tRNA cassettes to be used for each application, only one tRNA cassette is recommended when screening different tRNAs in order to facilitate cloning procedures. When screening either different ncAARS or the incorporation efficiency of different ncAAs by the same orthogonal pair, four tandem repeats of the tRNA cassette are preferred to achieve the highest yields of ncAA incorporation.
The optimized system for Azi incorporation including the humanized E2AziRS gene was deployed to map the binding pocket of the CRF1R and define binding paths of 5 different ligands: the two peptide agonists Ucn1 and CRF, and the three peptide antagonists Ucn1(8-40), CRF (9-41) and dFXCRF(12-41) (Figure 3). While the efficiency of Azi incorporation was previously shown to decrease when approaching the GPCR C-terminus33,34, our optimized system for Azi incorporation enables comparable expression levels of Azi-mutants with TAG-sites in different parts of the GPCR gene (Figure 4A). The results in Figure 4B show the screening of the extracellular loop 2 (ECL2) and helix V of CRF1R as a representative part of the ligand-binding pocket. A band at the correct size of the crosslinking product reveals that the position lies in the proximity of the ligand within the ligand-receptor complex, i.e. it is part of the binding pocket. Multiple crosslinking hits were found with all ligands tested, revealing distinct binding patterns for the peptide agonists and the antagonists. Notably, the pattern of crosslinking hits provides information about structural elements of the receptor. A number of successive hits, as observed in the ECL2 (positions F260-R263 in combination with antagonists) suggests a flexible loop region. A pattern of hits every three to four residues as found in helix V (D269/Y270, Y272/Q273, I277) hints towards a helical structure. The screen can be extended to all relevant domains of the GPCR. If a 3D structure or a molecular model of the receptor exists, ligand footprints can be visualized by highlighting the crosslinking hits for each ligand (Figure 5).
Both fluorescence and Western blot data (Figure 2C) suggest that TCO*K is the ncAA for bioorthogonal chemistry that gets incorporated with the highest efficiency by the MbPylRSAF. Different PylRS mutants might give higher incorporation yields of other click ncAAs17, but they were not tested in this study. TCO*K was incorporated into a CRF1R-EGFP fusion protein and enabled installing via SPIEDAC chemistry (Figure 6A) a small bright fluorophore on the receptor (Figure 6B). As a proof of specific labeling, the fluorescence of the label should be visible only in cells expressing the receptor (green cells in Figure 6B) and not in dark cells, which thus provide an internal negative control in each experiment. Receptors labeled using ultrafast SPIEDAC chemistry are still functional. After adding a peptide agonist, fluorescent compartments were observed throughout the cytosol, revealing the physiological process of GPCR internalization (Figure 6C). The signals of the fused EGFP co-localized with the fluorescent dye at all times, confirming the selective biorthogonal labeling of the GPCR.
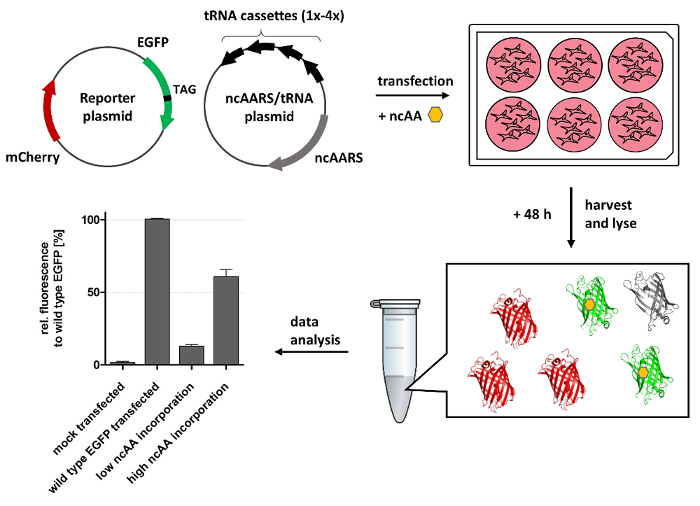
Figure 1: Fluorescence-based assay to evaluate the efficiency of stop codon suppression. HEK293 cells are co-transfected with two plasmids in the presence of the ncAA. One plasmid encodes for the desired ncAARS and suppressor-tRNA. The second plasmid encodes for the EGFP gene bearing a TAG stop codon at a permissive site together with a mCherry control. Two days after transfection, the green and red fluorescence of whole-cell lysates are measured in a plate reader. As the EGFP N-segment upstream the stop codon (grey) is not fluorescent, the yield of full-length EGFP (green) directly correlates to the efficiency of ncAA incorporation, while mCherry (red) provides an independent reference for normalization. The efficiency of the orthogonal system is given by the ratio between the amount of EGFP obtained via stop codon suppression and the amount of wild-type EGFP obtained by regular translation (no amber suppression). The figure is modified from Serfling, R. & Coin, I; Incorporation of Unnatural Amino Acids into Proteins Expressed in Mammalian Cells, Methods in Enzymology, 2016, 580, 89-107.29 Reproduction was permitted by the Copyright Clearance Center of Elsevier. Please click here to view a larger version of this figure.
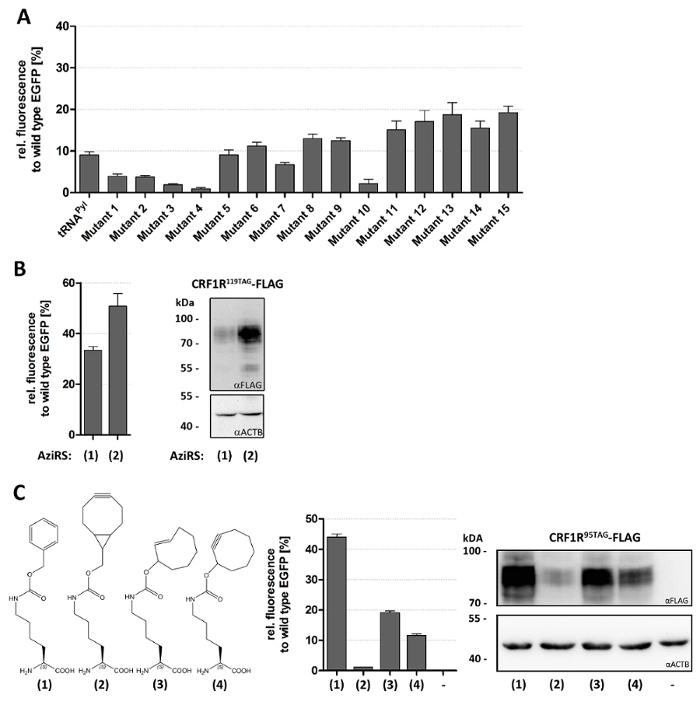
Figure 2: Representative applications of the fluorescence assay. (A) Screening of tRNAPyl variants30. HEK293 cells were co-transfected with the reporter plasmid and plasmids encoding for the MbPylRS/tRNA pair (one copy tRNA). Bars represent the relative fluorescence of EGFP obtained from the incorporation of Lys(Boc) compared to wild-type EGFP. (B) Evaluating the influence of codon-usage for ncAARS genes. (left panel) Fluorescence assay of cells transfected with two different gene variants of E2AziRS. (1) codon usage from E. coli and (2) humanized gene. (right panel) Western blot analysis of CRF1R95Azi-FLAG. The full-length receptor is detected by an anti-FLAG antibody. Actin β was used as loading control. (C) Evaluating the incorporation efficiency of three ncAAs designed for bioorthogonal chemistry. (left panel) Structures of ncAAs used in this experiment. (1) LysZ, which was used as positive control, (2) BCNK, (3) TCO*K and (4) SCOK. (central panel) Fluorescence assay of HEK293 cells transfected with a plasmid encoding for MbPylRSAF and four copies of tRNA15 from (A) to incorporate (1), (2), (3) and (4). (right panel) Western blot analysis of a CRF1R-FLAG mutant bearing one of the four ncAAs at position 95. Actin β was used as loading control. Panel A was adapted from Serfling, R. et al. Designer tRNAs for efficient incorporation of non-canonical amino acids by the pyrrolysine system in mammalian cells Nucleic Acids Res. 46 (1), 1-10 (2018) and are reproduced according to the Creative Commons Attribution license. Please click here to view a larger version of this figure.
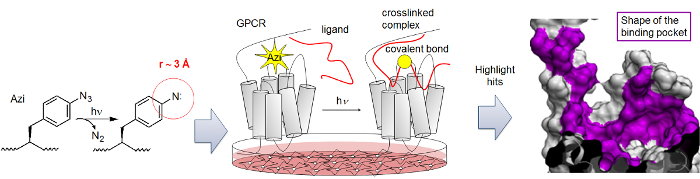
Figure 3: Schematic representation of the Azi-mediated photo-crosslinking mapping. A photo-activatable azido function (yellow star) is placed into a defined position within the receptor (grey). When the azido group is located proximal to the bound ligand (red), a covalent crosslinking product is formed upon UV irradiation (yellow circle indicates crosslinking site). The incorporation of the azido group into different positions of the receptor reveals the binding surface (purple) of the ligand, which represents the binding pocket. Please click here to view a larger version of this figure.
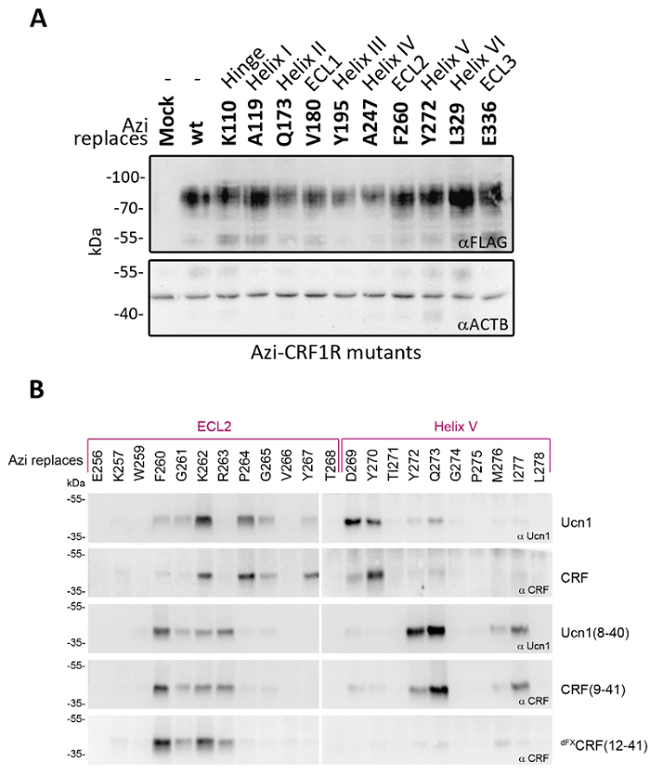
Figure 4: Incorporation of Azi throughout CRF1R for photo-crosslinking mapping of the binding pocket. (A) Comparison of expression levels of Azi-CRF1R-FLAG mutants towards wild-type receptor. The Azi incorporation sites are distributed throughout the whole receptor as indicated in the top row. Anti-FLAG Western blots of whole-cell lysates are shown. Actin β was used as loading control. (B) Western blots probed with either anti-CRF or anti-Ucn1 antibodies are shown. The residues replaced by Azi are indicated in the upper row. The cells were incubated with the ligands listed on the right, followed by UV irradiation at 365 nm and lysis. The samples were resolved on 10 % SDS-PAGE, deglycosylated using PNGase F and analyzed by Western blotting. The deglycosylated ligand-CRF1R complex runs at an apparent MW of ~40 kDa33. The non-crosslinked ligand is not detected (MW ~3-4 kDa). Both panels of this figure have been modified from Seidel, L. et al. Structural insight into the activation of a class B G-protein-coupled receptor by peptide hormones in live human cells. Elife.6 10.7554/eLife.27711 and are reproduced according to the Creative Commons Attribution license. Please click here to view a larger version of this figure.
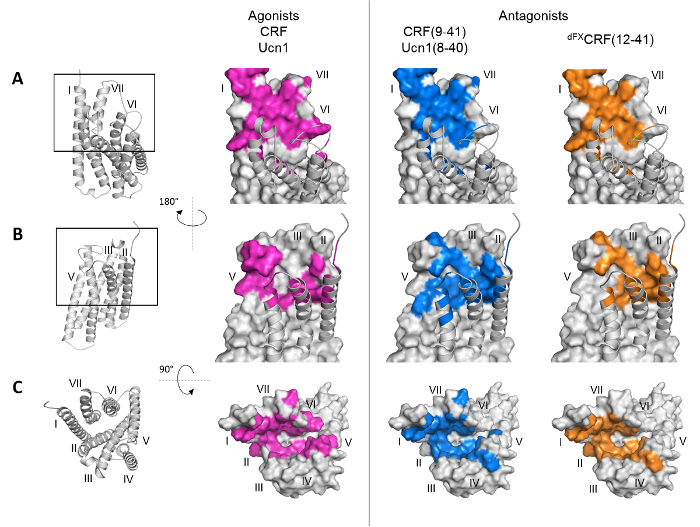
Figure 5: Footprints of peptide agonists and antagonists on the class B GPCR CRF1R. Surface representation of the CRF1R transmembrane domain. Positions of CRF1R that crosslinked the ligand when substituted by Azi are highlighted. Footprints of the peptide agonists CRF and Ucn1 are highlighted in magenta, footprints of the antagonists CRF (9-41) and Ucn1(8-40) in blue. The footprint of the antagonist dFXCRF(12-41) is highlighted in orange. The seven transmembrane helices I-VII are indicated by roman numbers. (A, B) Side views of the binding pocket from the membrane plane. (C) Top view into the binding pocket from the extracellular side. Reprinted from Seidel, L. et al. Structural insight into the activation of a class B G-protein-coupled receptor by peptide hormones in live human cells. Elife.6 10.7554/eLife.27711 and are reproduced according to the Creative Commons Attribution license. Please click here to view a larger version of this figure.
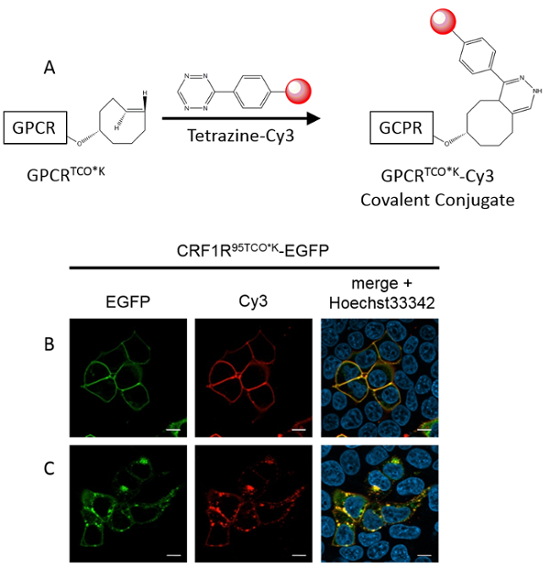
Figure 6: SPIEDAC labeling of CRF1R on live cells. (A) Reaction scheme of the strain-promoted inverse electron-demand Diels-Alder cycloaddition (SPIEDAC): the trans-cyclo-2-octene group of the ncAA TCO*K reacts with the tetrazine group linked to the fluorophore Cy3. (B, C) HEK293T cells expressing CRF1R95TCO*K-EGFP. Representative images of the green, red and blue channel. The size of the scale bar is 10 µm. (B) Cells were treated with tetrazine-Cy3 (1.5 µM) for 5 min. Only cells expressing the receptor (green) show occurrence of labeling (red). (C) Cells were treated with 200 nM Ucn1 and imaged after 15 min. Intracellular vesicles correspond to the internalized receptor. Panels B and C are modified from Serfling, R. et al. Designer tRNAs for efficient incorporation of non-canonical amino acids by the pyrrolysine system in mammalian cells Nucleic Acid Res. 46 (1) 1-10 (2018) (Oxford University Press) and are reproduced according to the Creative Commons Attribution license. Please click here to view a larger version of this figure.
