Following the flow chart of the Fab library construction (see Figure 1), we prepared M13KO7 helper phage pre-infected E. coli SS320 electro-competent cells. The efficiency of these electro-competent cells is estimated as 2 X 109 cfu/µg when the Fab phagemid backbone for library construction was used (Figure 4).
The uracil incorporation efficiency by comparison of titer in both E. coli CJ236 and E. coli SS320 cells was checked. The E. coli CJ236 and E. coli SS320 cells were infected by phage harboring dU-ssDNA. E. coli SS320 has enzymes (dUTPase and uracil glycosylase) that can degrade uracil-containing DNA, while E. coli CJ236 lacks these enzymes and cannot degrade uracil-containing DNA. To achieve an acceptable uracil incorporation efficiency, titers from E. coli CJ236 need to be at least 104 times higher than those from E. coli SS320. Otherwise the wild-type population will increase in the constructed antibody library due to inefficient uracil incorporation. Figure 5 showed that the titer from E. coli CJ236 is approximately 3 X 105 times higher than that from E. coli SS320, indicating an efficient uracil incorporation into phage ssDNA.
Next, we prepared and extracted dU-ssDNA. The dU-ssDNA purity is checked by agarose gel electrophoresis (Figure 6). Then the oligonucleotide-directed mutagenesis was conducted and the efficiency of the dU-ssDNA conversion to CCC-DNA was evaluated (Figure 6). Three products with lower motility than dU-ssDNA can be visualized on the gel including the fastest-running band (CCC-dsDNA), the middle weak band (nicked band), and the slowest-running band (strand-displaced DNA).
After electroporation into E. coli SS320, the library size was estimated from the overnight incubation plate (see step 4.7.5). The average library size was 5 X 109 from duplicate serial dilutions on LB/carb plates (Figure 7). However, the estimated size at this step may contain phage that do not display Fabs due to the presence of a frameshift or stop codon, or display misfolded Fabs. Sequencing and ELISA were used to estimate the functional diversity of constructed library. 96 randomly picked single clones were sent for sequencing analysis. Table 4 shows that 90 out of the 96 randomly picked single clones were successfully sequenced, which contains 70 clones without a premature stop codon (53 clones with at least one CDR mutant and 17 clones with the wild-type sequence) and 20 clones with a premature stop codon at different regions. Within the 70 clones, mutant rates of CDRH1, CDRH2, CDRH3, and CDRL3 are 50%, 57%, 53%, and 56%, respectively, while the mutant rate with at least one CDR is 76%. In the 20 clones with a premature stop codon (90%), the premature stop codon was mainly derived from the frameshift of oligonucleotide mutagenesis primers, including 45% (CDRH1), 10% (CDRH2), 15% (CDRH3), and 20% (CDRL3).
To detect the display of properly folded Fabs, a protein A/L based ELISA was employed as it is known that protein A and protein L can recognize proper folding of the VH framework and VL framework, respectively17,18. In agreement with the sequencing analysis, the ELISA assay in triplicate (Figure 8) showed that the 20 clones with a premature stop codon were all negative while the 17 clones with a wild-type sequence were all positive when the positive ratio was empirically set at 3.0. For the 53 clones with at least one CDR mutant, 43 clones were positive in ELISA while 10 clones were negative; this indicates that most of the clones were well folded while the CDRs from the 10 clones can have detrimental effects on Fab folding. In total, 43 clones out of the 90 clones (48%) were well folded and contained at least one CDR mutant. Thus, the functional amino acid diversity of the constructed library based on protein A/L ELISA and sequence analysis was estimated to be 2.4 X 109 (i.e., 48% of 5 X 109).
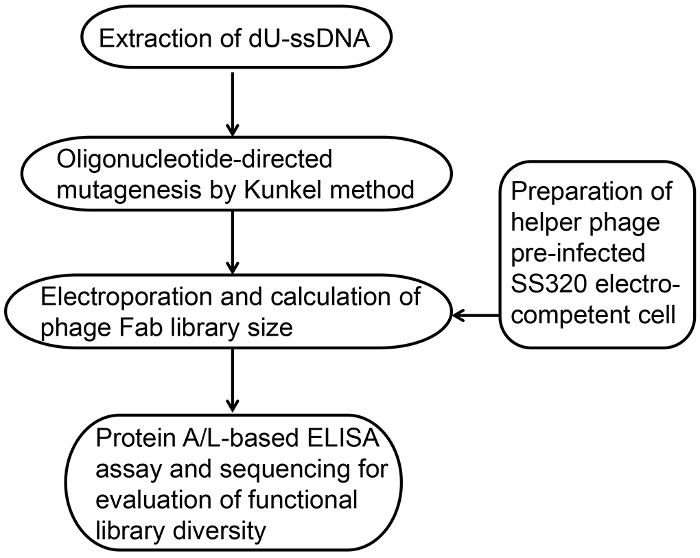
Figure 1: Overview of the phage-displayed Fab library construction. Phage-displayed Fab library construction follows a basic series of steps. It involves preparation of high-efficiency electro-competent bacterial cells, extraction of dU-ssDNA, Kunkel's method based oligonucleotide-directed mutagenesis, electroporation and calculation of phage Fab library size, functional evaluation by protein A/L ELISA, and DNA sequence analysis. Please click here to view a larger version of this figure.
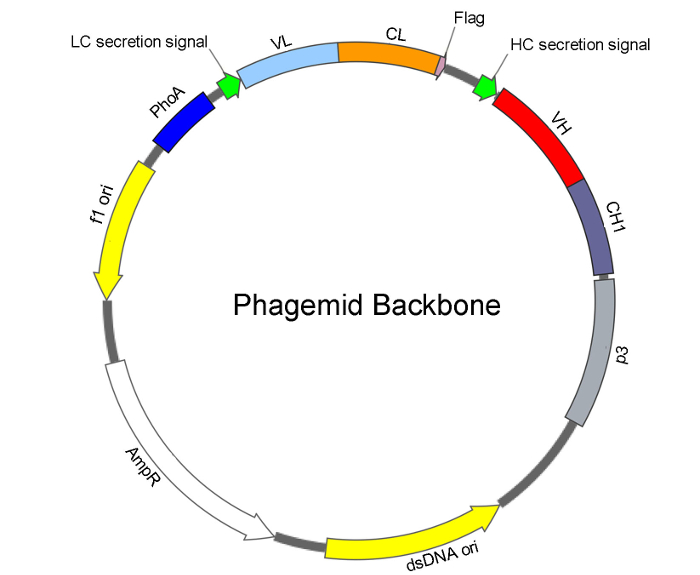
Figure 2: Phagemid architecture for the Fab library construction. The basic features of the phagemid backbone consist of origins of single-stranded (f1 ori) and double-stranded (dsDNA ori) DNA replication, and an ampicillin/carbenicillin resistance gene (AmpR). For Fab display, under the control of the alkaline phosphatase promoter (PhoA), the phagemid contains a bicistronic cassette to drive expression and secretion of: light chain (LC) consisting of a secretion signal, VL (variable region of light chain), CL (constant region of light chain), and C-terminal flag tag; and heavy chain (HC) consisting of a secretion signal, VH (variable region of heavy chain), and CH1 (constant region 1 of heavy chain) fused with a p3 phage minor coat protein. Assembly of the light chain and heavy chain into Fab within the E. coli periplasm directs the display of Fab on the phage surface. Please click here to view a larger version of this figure.
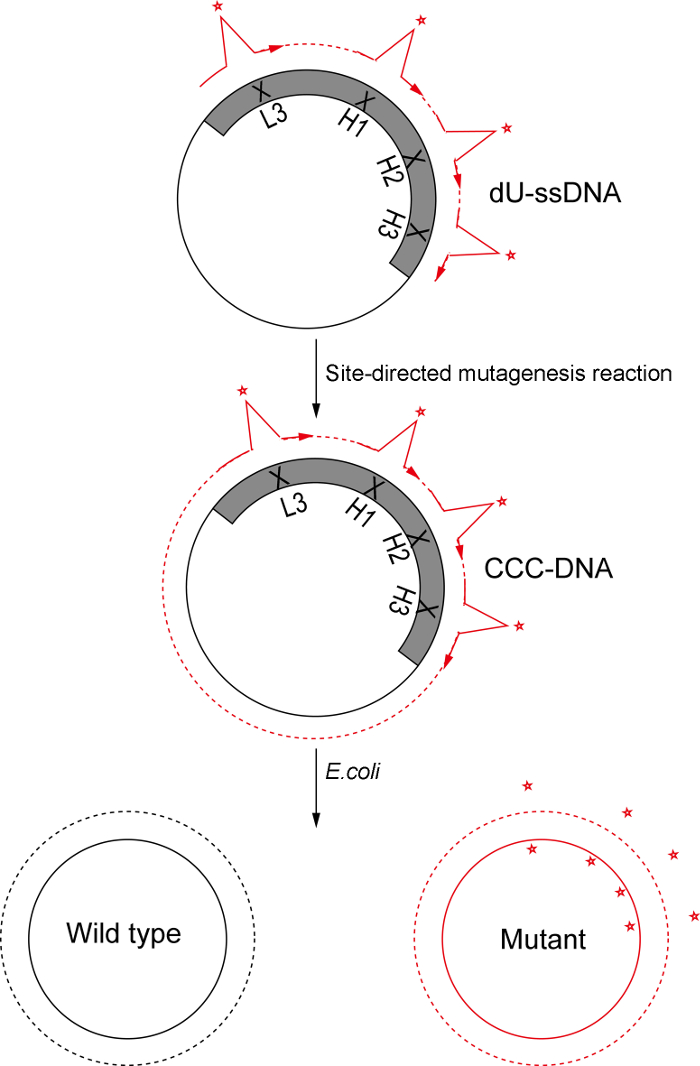
Figure 3: Schematic of Kunkel's method based oligonucleotide-directed mutagenesis. In this protocol, we used Kunkel's method to prepare dU-ssDNA template. Oligonucleotides for CDRH1, CDRH2, CDRH3, and CDRL3 with designed diversity are phosphorylated, annealed to the template, and used to convert ss-DNA to CCC-dsDNA. Following electroporation into E. coli SS320 electro-competent cells, the heteroduplex DNA is repaired to either the wild type or the mutant form; Due to the presence of uracil in the wild type strand, the repair process favors the mutant form, and thus, the mutant form dominates the library. Please click here to view a larger version of this figure.
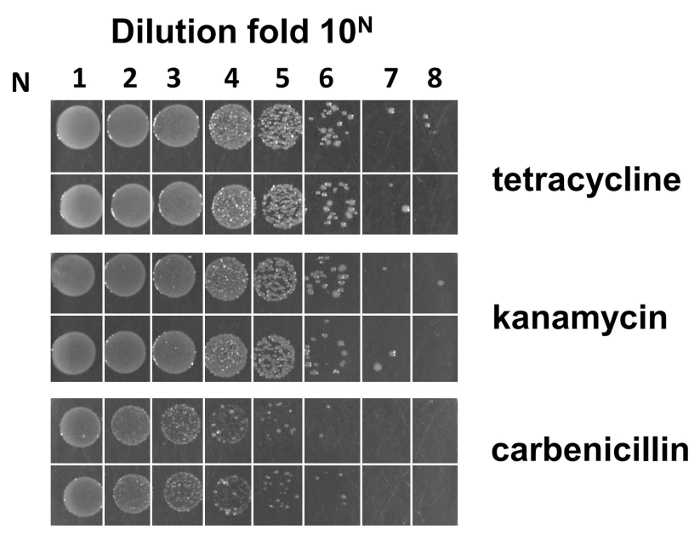
Figure 4: Estimation of the M13KO7 pre-infected E. coli SS320 electro-competent cell efficiency. A phagemid backbone vector was used to check the electroporation efficiency of the competent cells. Formula to calculate the efficiency is as follows: assume that M is the average colony number counted from the most diluted fold 10N (N is from 1-8) in duplicate. E. coli SS320 efficiency from LB/carb plate is equal to M X 10N+3 cfu/µg. The efficiency of the electro-competent cells is around 2 X 109 cfu/µg. Please click here to view a larger version of this figure.
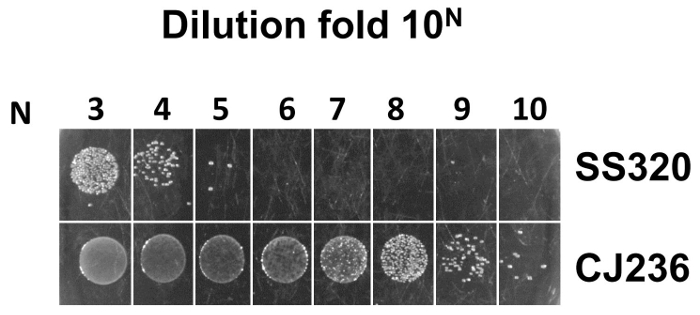
Figure 5: Assessment of uracil incorporation into ssDNA by phage infection of E. coli CJ236 and E. coli SS320 cells. Based on Kunkel's method, the uracil incorporation efficiency is checked by comparison of phage infection titer in both E. coli CJ236 and E. coli SS320 cells. The titer calculation formula is as follows: assume that M is the average colony number counted from the most diluted fold 10N (N is from 1-10), and that the titer from E. coli CJ236 or E. coli SS320 is equal to M X 10N+2 cfu/mL. The efficiency of uracil incorporation can be estimated from the titer ratio of E. coli CJ236 and E. coli SS320. The titer in E. coli CJ236 was 9 X 1012 cfu/mL while the titer in E. coli SS320 was 3 X 107 cfu/mL. The titer ratio of E. coli CJ236 and E. coli SS320 was 3 X 105. Please click here to view a larger version of this figure.
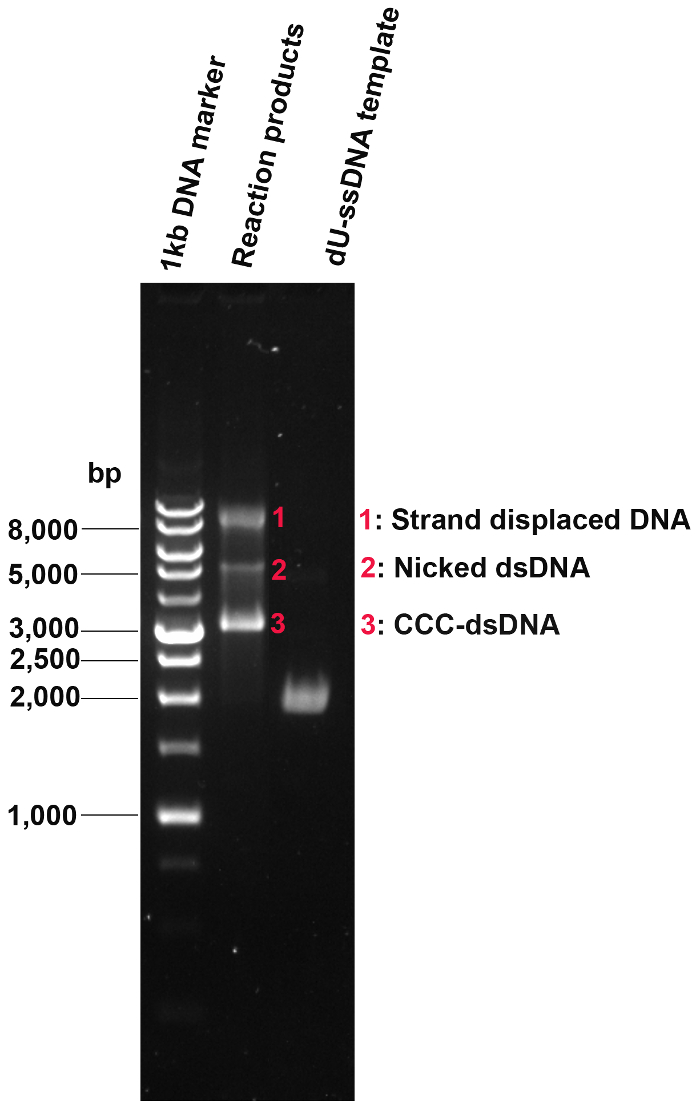
Figure 6: Conversion of dU-ssDNA to CCC-DNA by oligonucleotide-directed mutagenesis. Following oligonucleotide-directed mutagenesis, the efficiency of dU-ssDNA conversion to CCC-DNA was evaluated. dU-ssDNA was completely converted to dsDNA. The dominant band is CCC-dsDNA while there is a minor portion of nicked dsDNA and strand-displaced DNA. Please click here to view a larger version of this figure.
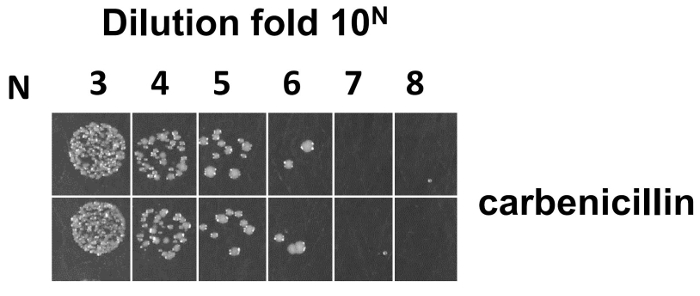
Figure 7: Phage titration for calculation of library size. After electroporation into E. coli SS320, the library size was estimated from serial dilutions on LB/carb plates. The size calculation formula is as follows: assume M is the average colony number counted from the most diluted fold 10N from a 2YT/Carb plate (N is from 1-8), size is equal to 2M X 10N+3. Please click here to view a larger version of this figure.
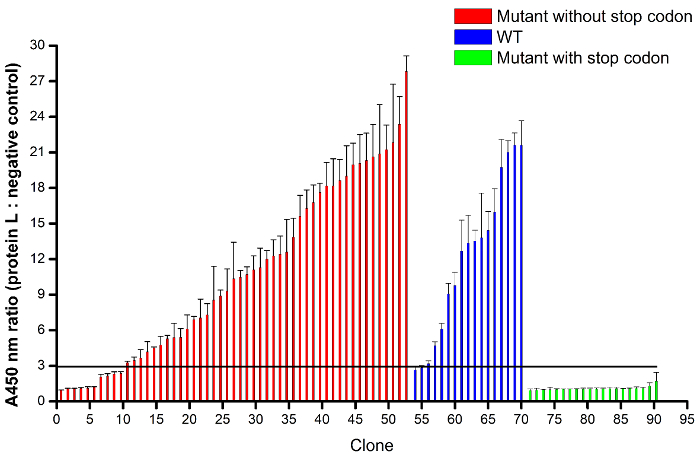
Figure 8: Protein A/L direct binding phage ELISA. Protein L can recognize the framework of well folded kappa light chain VL and protein A can recognize the framework of well folded heavy chain VH. Binding of Fab with protein L and A indicates proper folding of both heavy chain and light chain. In brief, protein L in triplicate and the negative control M-PBST were coated to the plate, Fab phage supernatants from different clones were incubated with protein L and M-PBST, then after wash, protein A-HRP was used to capture bound Fab phage. Phage ELISA readings showed 90 randomly picked clones with successful sequencing readout. A threshold line representing the clone as positive was empirically defined where the ratio of OD450 absorbance value from protein L (average of triplicate with error bar) versus negative control was more than 3.0. Three groups based on sequencing analysis were shown, corresponding to a mutant without stop codon in red (53 clones), wild type (WT) in blue (17 clones), and mutant with stop codon in green (20 clones). Please click here to view a larger version of this figure.
| Reagent setup | Component | Amount | comments/description |
| 2YT medium | Yeast extract | 10 g | Add ultrapure water to make up the volume to 1.0 L, adjust pH to 7.0, autoclave. |
| Tryptone | 16 g | ||
| NaCl | 5 g | ||
| 2YT top agar | Yeast extract | 10 g | Add ultrapure water to make up the volume to 1.0 L and adjust pH to 7.0, heat to dissolve, autoclave. |
| Tryptone | 16 g | ||
| NaCl | 5 g | ||
| Granulated agar | 7.5 g | ||
| 2YT/carb/cmp medium | Carbenicillin | 100 μg/mL | |
| Chloramphenicol | 10 μg/mL | ||
| 2YT/carb/kan/uridine medium | Carbenicillin | 100 μg/mL | |
| Kanamycin | 50 μg/mL | ||
| Uridine | 0.25 μg/mL | ||
| 2YT/carb/tet medium | Carbenicillin | 100 μg/mL | |
| Tetracycline | 10 μg/mL | ||
| 2YT/carb medium | Carbenicilin | 100 μg/mL | |
| 2YT/kan medium | Kanamycin | 50 μg/mL | |
| 2YT/kan/tet medium | Kanamycin | 50 μg/mL | |
| Tetracycline | 10 μg/mL | ||
| 2YT/tet medium | Tetracycline | 10 μg/mL | |
| 2YT/cmp medium | Chloramphenicol | 10 μg/mL | |
| LB medium agar | Yeast extract | 5 g | Add ultrapure water to make up the volume to 1.0 L, adjust pH to 7.0, autoclave. For LB agar, add 20 g of granulated agar, autoclave. |
| Tryptone | 10 g | ||
| NaCl | 10 g | ||
| LB/carb plates | LB agar | 1L | |
| Carbenicillin | 100 μg/mL | ||
| LB/tet plates | LB agar | 1 L | |
| Tetracycline | 10 μg/mL | ||
| LB/cmp plates | Chloramphenicol | 10 μg/mL | |
| LB/kan plates | Kanamycin | 50 μg/mL | |
| SOC medium | Yeast extract | 5 g | Add ultrapure water to make up the volume to 1.0 L and adjust pH to 7.0, autoclave. |
| Tryptone | 20 g | ||
| NaCl | 0.5 g | ||
| KCl | 0.2 g | ||
| 2.0 M MgCl2 | 5.0 mL | ||
| 1.0 M glucose | 20 mL | ||
| Superbroth medium | Tryptone | 12 g | Add ultrapure water to 900 mL, autoclave, add 100 mL of autoclaved 0.17 M KH2PO4, 0.72 M K2HPO4. |
| Yeast extract | 24 g | ||
| Glycerol | 5 mL | ||
| Superbroth kan/tet medium | Kanamycin | 50 μg/mL | |
| Tetracycline | 10 μg/mL | ||
| 1X PBS | NaCl | 137 mM | Adjust pH to 7.2, autoclave. |
| KCl | 3 mM | ||
| Na2HPO4 | 8 mM | ||
| KH2PO4 | 1.5 mM | ||
| TAE/agarose gel | TAE buffer | ||
| Agarose | 1% (w/v) | ||
| GelRed | 1:10000 (v/v) | ||
| TMB substrate | TMB | 50% (v/v) | |
| H2O2 peroxidase substrate | 50% (v/v) | ||
| M-PBST buffer | 1X PBS | 100 ml | |
| Tween-20 | 0.05% (v/v) | ||
| NON-Fat Powdered Milk | 5% (v/v) | ||
| 5X PEG/NaCl | PEG-8000 | 20% (w/v) | Add ultrapure water to make up the volume to 1L, and autoclave. |
| NaCl | 2.5 M | ||
| PBST buffer | 1X PBS | 1 L | 0.22 μm filter-sterilize. |
| Tween-20 | 0.05% (v/v) | ||
| 10X TM buffer | MgCl2 | 0.1 M | Adjust pH to 7.5. |
| Tris | 0.5 M | ||
| 1.0 mM HEPES, pH 7.4 | 1.0 M HEPES | 4.0 mL | 0.22 μm filter-sterilize. |
| Ultrapure water | 4.0 L | ||
| 10% (v/v) ultrapure glycerol | Ultrapure glycerol | 100 ml | 0.22 μm filter-sterilize. |
| Ultrapure water | 900 mL | ||
| Ultrapure water | H20 | Dnase-free, Rnase-free, Pyrogen-free. |
Table 1: Reagent setup.
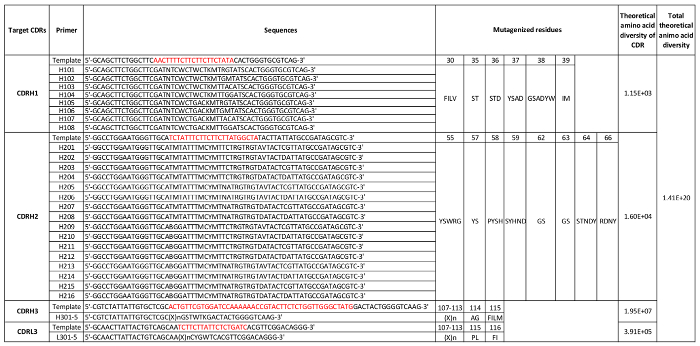
Table 2: CDR diversities and mutagenesis primers. The DNA sequences of the CDR regions to be mutated are shown in red; sequences are formatted using the IUPAC nucleotide code. "X" indicates tri-nucleotide from a mixture designed to contain different amino acid sets; "n" indicates different number of X. Five primers with a different number of X were used to diversify CDRL3 or CDRH3, respectively, to generate variable length of CDRL3 and CDRH3. The residue numbers are defined by the IMGT nomenclature. Please click here to download this table.
| Kunkel's method based mutagenesis | ||
| Reaction 1. Oligonucleotide phosphorylation with T4 polynucleotide kinase | ||
| Component | Amount | Final |
| mutagenic oligonucleotides | 0.6 μg | |
| 10X TM buffer | 2 μL | 1X |
| 10 mM ATP | 2 μL | 1 mM |
| 100 mM DTT | 1 μL | 5 mM |
| T4 polynucleotide kinase (10 U/μL) | 2 μL | 20 U |
| Ultrapure H20 | Up to 20 μL | |
| Reaction setting | ||
| Step 1. | 37 °C for 1 h | |
| Reaction 2. Annealing of the oligonucleotides to the template | ||
| Component | Amount | Final |
| dU-ssDNA template | 20 μg | 20 μg |
| 10X TM buffer | 25 μL | 1X |
| phosphorylated CDRH1 oligonucleotides | 20 μL | 0.6 μg |
| phosphorylated CDRH2 oligonucleotides | 20 μL | 0.6 μg |
| phosphorylated CDRH3 oligonucleotides | 20 μL | 0.6 μg |
| phosphorylated CDRL3 oligonucleotides | 20 μL | 0.6 μg |
| Ultrapure H20 | Up to 250 μL | |
| Reaction setting | ||
| Step 1. | 90 °C for 3 min | |
| Step 2. | 50 °C for 5 min | |
| Step 3. | 20 °C for 5 min | |
| Reaction 3. Enzymatic synthesis of CCC-dsDNA | ||
| Component | Amount | Final |
| annealed oligonucleotides/template mixtures | 250 μL | |
| 10 mM ATP | 10 μL | 346 µM of each nucleotide |
| dNTP mix (25 mM of each nucleotide) | 10 μL | 865 µM of each nucleotide |
| 100 mM DTT | 15 μL | 5 mM |
| T4 DNA ligase | 1 μL | 30 Weiss units |
| T7 DNA polymerase | 3 μL | 30 U |
| Reaction setting | ||
| Step 1. | 20 °C for overnight | |
Table 3: Procedures and components of Kunkel's method based reaction.
| Group | Clone number | Region | Percentage | ||
| No premature stop codon | 70 | CDRH1 mutation | 50% (35/70) | ||
| CDRH2 mutation | 57% (40/70) | ||||
| CDRH3 mutation | 53% (37/70) | ||||
| CDRL3 mutation | 56% (39/70) | ||||
| At least one CDR mutation | 76% (53/70) | ||||
| Premature stop codon | 20 | CDRH1 defect | 45% (9/20) | ||
| CDRH2 defect | 10% (2/20) | ||||
| CDRH3 defect | 15% (3/20) | ||||
| CDRL3 defect | 20% (4/20) | ||||
| Other defect | 10% (2/20) | ||||
Table 4: Sequence analysis of CDRH1, CDRH2, CDRH3, and CDRL3 from the synthetic Fab library.
