Epilepsy is a life-threatening progressive disorder causing uncontrolled activity of the brain1; it carries among the highest burden of disease and significant social stigma2,3. TLE is the most frequent syndrome (40%) and the most frequently (~ 30%) resistant to anti-epileptic medications4. While surgical ablation of the epileptogenic tissue may ameliorate the patient's condition, it is not feasible in all patients and may not guarantee a completely seizure-free life5. Modulation of epileptic limbic networks by electrical DBS is a promising approach when pharmacological treatment or neurosurgery are not suitable.
Rodent brain slices are a valuable tool to study how neuronal networks function in health and disease6 by means of in vitro electrophysiology techniques, as they preserve, at least in part, the original architecture and connectivity of the brain region(s) of interest (ROI). In particular, horizontal combined hippocampus-entorhinal cortex (hippocampus-EC) slices comprise the essential neuronal networks involved in TLE and are therefore routinely employed in in vitro TLE research7.
Seizure-like activity may be acutely induced in brain slices by changing the ionic composition of the artificial cerebrospinal fluid (ACSF), such as decreasing magnesium while increasing potassium8,9,10, or by means of pharmacological manipulations, such as blocking inhibitory GABAergic activity (see11 for a comprehensive review). However, these models are based on the unbalanced alteration of excitation and inhibition; thus, they do not allow studying the interaction and concerted contribution of excitatory and inhibitory networks to ictogenesis. Continuous perfusion of brain slices with the convulsant drug 4AP enhances both excitatory and inhibitory neurotransmission, thus allowing to study acute ictogenesis while keeping synaptic activity overall intact12.
MEAs allow recording the electrical activity generated by neuronal networks from a greater number of observation points as compared to conventional extracellular field potential recording, where spatial restrictions limit the number of electrodes that can be accommodated on the brain slice surface. Moreover, MEA chips offer the added advantage of known inter-electrode distance, which is very useful to trace propagation and assess the traveling velocity of recorded signals. Although initially conceived for recording from cultured neurons13,14, MEAs are now also used to characterize the electrophysiological features of acute brain slices obtained from rodents15 and humans16. Thus, in the context of epilepsy research, MEAs represent a valuable tool to pinpoint neuronal networks' interactions at the core of ictogenesis16,17,18.
However, MEA recording inherently carries technical challenges in obtaining or maintaining a stable epileptiform pattern throughout long (several hours) experimental protocols. First, tissue oxygenation may not be adequate within the large and round submerged-type recording chamber19,20 typical of commercially available MEAs, while poor signal-to-noise ratio and temperature instability might affect the quality of the recording when a high perfusion rate (6 – 10 mL/min) is used to improve the oxygen supply to the brain slice (see for example the technical notes on heating and perfusion equipment21). Second, recurrent discharges featuring high frequency components, such as epileptiform discharges, are hardly observed when using submerged-type recording chambers19; this is particularly the case when the acute epileptiform pattern is chemically induced and involves ephaptic mechanisms, as it is the case for the 4AP model22 (see11 for a comprehensive overview). To overcome these limitations, several strategies have been proposed by researchers. For example, increasing the perfusion rate19,20 while reducing brain slice thickness (≤300 µM) and area17,18 are crucial factors to achieving adequate oxygen supply to the tissue. In addition, improved brain slice viability can also be accomplished by using perforated MEAs (pMEAs), which allow perfusing the brain tissue from both sides18.
Whereas the described approaches have significantly improved the feasibility of MEA recording from brain slices, they have not been tested against prolonged (several hours) recording and electrical stimulation sessions, the latter representing a significant stress for the brain tissue. Prolonged recording sessions may be required to study the evolution in time of specific features of epileptiform patterns that would not be possible to unmask by short-term measurements. In the context of DBS research, prolonged experimental protocols may be required to evaluate and compare the effects of several stimulation paradigms in the same brain slice.
When only spontaneous electrical activity needs to be recorded, mapping of the electrodes with respect to the ROI is usually done a posteriori, i.e., during data analysis; instead, the study of evoked responses or of electrical neuromodulation paradigms requires that stimulation be delivered to specific ROI(s), dictating the need of quick and easy live electrode mapping during the experiment.
Here, we illustrate a simple experimental protocol that allows for induction and maintenance of a stable 4AP-induced epileptiform pattern in adult rodent brain slices. The observed activity faithfully reproduces the electrophysiological features of this model as characterized using conventional extracellular electrophysiology techniques. It persists for several hours and outlasts sustained electrical stimulation for repeated prolonged epochs. The chambers used to maintain and incubate the brain slices can be easily assembled using standard laboratory supplies (Figure 1A), whereas a custom recording chamber allowing for optimal solution exchange rate and laminar flow (Figure 1B) can be obtained from commercial sources or using 3D printing technology at an affordable price. Quick mapping of the ROI(s) for real-time monitoring and for the selection of stimulating electrodes is made possible by a custom user-friendly GUI named mapMEA, freely available upon request.

Figure 1: Custom equipment used for this protocol. (A) The holding chambers for recovery, pre-warming, and pre-incubation in 4AP are assembled using a beaker and a Petri dish. The Petri dish should be smaller in diameter than the breaker and is held in place by means of a syringe plunger. The bottom of the Petri dish is replaced by a nylon mesh (from soft stockings) glued with cyanoacrylate onto the rim of the Petri dish. Oxygen is supplied via a bent spinal needle (22 G), inserted between the walls of the Petri dish and the beaker. Bubbles should be emerging from the side of the beaker and never reach the nylon mesh to avoid brain slice displacement. The top of a Petri dish can be used as a lid to avoid ACSF evaporation and maintain oxygen saturation. (B) Custom recording chamber. in: inlet reservoir to accommodate the heating cannula and the reference electrode. out: outlet reservoir to accommodate the suction needle. rec:recording chamber. (C) Glass Pasteur pipette, curled by fire-polishing, used to handle the tissue and adjust its position within the recording chamber. (D) Custom slice hold-down anchor. (E) Final assembly of the recording chamber mounted onto the MEA chip. A brain slice rests onto the bottom held in place by the anchor. The red arrow indicates the PTFE tubing covering the heating cannula, whereas the red circle indicates the reference electrode, a saturated KCl pellet submerged by ACSF in the inlet reservoir. The blue arrow indicates the suction needle in the outlet reservoir. Please click here to view a larger version of this figure.
All methods described here have been approved by the Italian Ministry of Health (authorization n. 860/2015-PR) in compliance with the EU directive 2010/63/EC on animal welfare.
1. Preparation of the MEA/Custom Chamber Assembly
Note: Start 2 days before the experiment. Use a MEA without a ring (see Materials Table).
- Clean the MEA (duration: 35 – 40 min).
- Dissolve an enzymatic cleaner powder into warm distilled water. Brush the warm cleaning solution onto the MEA surface then soak the MEA into the warm cleaning solution for at least 3 h.
- Rinse the MEA thoroughly with distilled water and dry it using a lint-free no-scratch tissue.
- Assemble the MEA with the recording chamber (duration: 10 min + overnight).
- Evenly spread the elastomeric sealant on the bottom surface of the recording chamber. Make sure there are no bubbles.
- Mount the recording chamber onto the MEA with the help of tweezers and apply gentle pressure. If there are any bubbles, slightly move the chamber in a circle while applying gentle pressure with fingers until all the bubbles have disappeared. Spread a sealant layer all the way around the outer border of the recording chamber.
Note: CRITICAL! Do not cover the MEA contacts with the sealant. - Place the MEA/chamber assembly inside a 15 cm Petri dish. Place a 5 mL Petri dish or a 10 mL beaker filled with distilled water close to the MEA. Cover everything to maintain a moist environment. Let cure at room temperature overnight.
NOTE: The protocol can be paused here.
- Coat the MEA to make it hydrophilic (duration: 5 – 10 min + overnight).
- Place the MEA into a 15 cm Petri dish. Pour 50 µL of poly-D-lysine onto the MEA recording area. Close the Petri dish, seal it with sealing film, and store overnight at 4 °C.
- Rinse the MEA thoroughly using distilled water and store the MEA at 4 °C in distilled water.
NOTE: The protocol can be paused here. Coated MEAs may be stored and reused for several experiments. Routinely cleaning and re-coating the MEA helps remove tissue debris and improve signal-to-noise ratio.
2. Preparation of Stock Solutions
Note: Start 1 day before the experiment (duration: ~2 h). Stock solutions help speed-up the experiment on the experimental day. They can be prepared in advance and stored. See Table 1 for concentration factor and composition. Refer to Table 2 for composition of final solutions.
- Dissolve the chemicals in distilled water at room temperature. Filter the stock solutions with a filter bottle (filter diameter: 0.22 µm).
- Store at 4 °C (see Table 1 for recommended maximum storage time).
NOTE: The protocol can be paused here.
3. Preparation of Agar
Note: Start 1 day before the experiment (duration: ~1 h).
- Pour 250 mL of distilled water into a 500 mL beaker and start stirring at 350 rpm. Add 5 g of agar and wait until it is completely dissolved.
- Heat the solution at 250 – 300 °C. Let the water evaporate until the solution is ~200 mL to yield a 2.5% w/v agar solution.
NOTE: The temperature should be high enough to let water evaporate without bubbling. - Pour the agar solution onto a 200 mL plastic square box of ~1.5 cm-high walls and let it cool down at room temperature until it becomes solid.
- Cover the box and store it at 4 °C. The agar block is good for several weeks.
NOTE: The protocol can be paused here.
4. Preparation of the Frozen Plane
Note: Start 1 day before the experiment.
- Fill a flat-bottom stainless steel bowl with distilled water up to 0.5 – 1 cm below the rim.
- Store at -20 °C overnight.
5. Preparation of Neuroprotective Cutting ACSF
Note: Start 1 day before the experiment or the same day of the experiment (see Table 1 and Table 2) (duration: 30 min).
- Pour 200 mL of distilled water in a 500 mL beaker and start stirring using a magnetic stirrer at 350 rpm.
- Add 50 mL of stock B and 50 mL of stock C (see Table 1).
- Add 3 mM pyruvic acid, 208 mM sucrose, 10 mM D-glucose, and 1 mM L-ascorbic acid.
NOTE: The actual volume of pyruvic acid depends on the density and purity and may vary with the batch. Always calculate the required volume when opening a new batch. - Cover with sealing film and let stir for 30 min.
- Transfer the solution to a 500 mL volumetric flask and add distilled water to fill the flask.
- Seal the volumetric flask with sealing film and gently invert 3 – 5 times until the solution is evenly clear.
- Transfer the cutting ACSF into a glass bottle with cap.
- Store the cutting ACSF at -80 °C for 20 – 30 min or at 4 °C overnight to cool down to 4 °C.
NOTE: The protocol can be paused here if overnight cooling is preferred. Otherwise the time required for cooling the cutting ACSF may be used to pursue step 6.
6. Preparation of the Holding ACSF
Note: Prepare the solution fresh on the day of the experiment (duration: 5 – 10 min). The holding ACSF will be used to rinse the brain slices from the cutting ACSF during the slicing procedure (see step 8.16) and for brain slice long-term storage and pre-warming. To rinse the brain slices, 100 mL of holding ACSF is sufficient. The holding and pre-warming chambers used in this protocol are custom-made and contain a volume of 300 mL and 100 mL, respectively (cf. Figure 1A). With this setting, 500 mL of holding ACSF is sufficient. Chambers obtained from commercial sources may contain a different volume, in which case the overall amount of holding ACSF to be prepared should be adjusted accordingly.
- Pour 200 mL of distilled water in a 500 mL volumetric flask.
- Add 50 mL of stock A, 50 mL of stock B, 6.5 mL of stock M, and 1 mM L-ascorbic acid. Gently agitate using rotating movements until L-ascorbic acid is completely dissolved.
- Add distilled water to reach the final volume, seal the volumetric flask with sealing film, and gently invert 3 – 5 times until the solution is evenly clear. Refer to Table 2 for the holding ACSF composition.
7. Preparation of the Experimental Benches
Note: This requires 15 min, plus 30 min to obtain a steady temperature of the warm bath.
- Recording bench
- Start the warm bath, set it to 32 °C, and cover it to maintain its temperature.
NOTE: The warm bath used in this protocol can be custom-made using a hard plastic box and an aquarium thermostat pre-set at the desired temperature. Steady desired temperature may be reached in ~30 min starting from room temperature. Since the pre-warming and incubating chambers sit at the bottom of the plastic box, the water level should be just enough to cover the thermostat so as to keep the chambers from floating. - Assemble the holding and the pre-warming chambers (cf. Figure 1A), and pour the holding ACSF into them. Keep the holding chamber at room temperature and place the pre-warming chamber inside the warm bath. Start bubbling the solutions with 95% O2/5% CO2 gas mixture and remove any trapped bubbles using an inverted Pasteur pipette. Keep covered.
- Start the warm bath, set it to 32 °C, and cover it to maintain its temperature.
- Slicing bench and vibratome
- Glue a small agar block onto the center of the specimen disc using cyanoacrylate glue.
- Put a 250 mL beaker into an ice bucket and pour cutting ACSF into it.
- Take two 100 mL beakers and pour 50 mL of holding ACSF into each. Leave the beakers at room temperature. These are the rinsing beakers.
- Start bubbling the solutions with a 95% O2/5% CO2 gas mixture.
- Mount the buffer tray onto the vibratome and surround it with crushed ice. Pour some ethanol on the crushed ice to aid in maintaining cold temperatures during the slicing procedure. After pouring ethanol, add more ice as needed.
NOTE: A temperature of 2 °C is optimal. - Place the bubbler inside the buffer tray, then assemble and mount the vibratome blade block.
NOTE: Make sure that the blade is mounted at clearance angle of ~10°. - Put crushed ice in a 500 mL beaker up to 2/3 of the volume and fill with distilled water.
- Take the frozen bowl out of the freezer, place it upside-down on the slicing bench, and cover it with a paper towel and a filter paper disc.
- Anesthesia bench
- Place a small bowl in an ice-filled tray and pour cutting ACSF in it. Start bubbling with a 95% O2/5% CO2 gas mixture.
8. Brain Slice Preparation and Maintenance
Note: This protocol makes use of adult (4 – 6 weeks-old) male CD1 mice, but other strains (e.g., c57/bl617,18) can be used. We also later compare the experimental output obtained using mouse brain slices with that yielded by brain slices obtained from male Sprague-Dawley rats of the same age. The following steps refer to the preparation of partially disconnected brain slices in which CA3-driven fast interictal-like activity is restrained to the hippocampus proper and cannot propagate to the parahippocampal cortices, as described in23. Hippocampus-cortex disconnection is a pre-requisite to pursue electrical modulation studies in using the hippocampus-EC brain slice preparation. The vibratome used refers to the model listed in the Materials Table. Other models may require a different procedure.
- Anesthetize the rodent using isoflurane 5% in a carbogen gas mixture delivered to an anesthesia induction chamber at 2 L/min.
- Under deep anesthesia (no reflexes in response to foot and paws pinch), extract the brain within 1 min following standard procedures as in23. Place the brain into the small bowl containing equilibrated ice-cold cutting ACSF and let chill for 90 – 120 s.
NOTE: Do not let the brain chill for too long, otherwise it may freeze. - Pour the ice-cold cutting ACSF into the buffer tray. Spread cutting ACSF onto the filter paper placed on the frozen bowl. Place the brain onto the filter paper and isolate the required brain tissue block by removing the cerebellum and cutting the frontal pole out straight.
- With the help of a bent spatula, glue the dorsal side of the brain onto the specimen disc with the cut frontal side facing the agar block and the occipital pole facing the vibratome blade. Use a thin layer of cyanoacrylate glue. Place the specimen disc in the buffer tray immediately and secure it.
- Adjust the sectioning range so as to move the vibratome blade all the way across the tissue up to the agar block. Adjust the specimen disc height to reach the blade level with the brain tissue block.
- Discard the tissue sections until the hippocampus is clearly visible (usually ~ 900 µm). Then, set a slice thickness of 400 µm and start slicing to retain the tissue sections. For each brain section split the two hemispheres and trim the unnecessary tissue to obtain two brain slices. As the buffer tray is heightened during subsequent sections, fill the buffer tray lodgment with iced distilled water (see step 7.2.7).
- Use an inverted Pasteur pipette to gently transfer the brain slices in the first rinsing beaker, empty the Pasteur pipette, and transfer the brain slices to the second rinsing beaker. Then, transfer the brain slices into the holding chamber and let them recover for at least 60 min.
NOTE: Usually it is possible to obtain 6 – 8 brain slices overall. The protocol may be paused here.
9. Preparation of ACSF Containing 4-AP (4AP-ACSF)
Note: This requires 10 min for preparation, plus 20 min of warming. CAUTION! 4-AP is a convulsant drug and it is toxic. Handle with gloves and avoid spilling.
- Pour 200 mL of distilled water in a 500 mL volumetric flask.
- Add 50 mL of stock A, 50 mL of stock B, 5 mL of stock M, and 1 mM L-ascorbic acid. Gently agitate using rotating movements until the L-ascorbic acid is completely dissolved.
- Add 500 µL of 4AP stock.
- Add distilled water to reach the final volume, seal the volumetric flask with sealing film, and gently invert it 3 – 5 times until the solution is evenly clear.
- Assemble the 4AP incubating chamber (cf. Figure 1A) and pour ~80 mL of 4AP-ACSF into it. Cover the chamber, transfer it to the warm bath, and start bubbling with a 95% O2/5% CO2 gas mixture. Remove any trapped bubbles using an inverted Pasteur pipette. Keep covered.
- Wait until the 4AP-ACSF temperature is 30 – 32 °C (~20 min).
NOTE: The protocol may be paused here if brain slices are still recovering.
10. Pre-warming and Incubation of Slices in 4AP (Duration: 90 min)
- Check that the pre-warming ACSF and the 4AP-ACSF temperature is 30 – 32 °C.
- Use an inverted glass Pasteur pipette to transfer 1 brain slice into the pre-warming chamber and let the tissue rest for 25 – 30 min. Then transfer the brain slice into the 4AP-ACSF incubating chamber and let it rest for 60 min.
11. Preparation of the MEA Set-up
Note: Start 15 min before recording.
- Transfer the remaining volume of 4AP-ACSF into a 500 mL Erlenmeyer flask.
- Place the Erlenmeyer flask on a shelf above the MEA amplifier and use tubing to allow the solution to continuously feed a 60 mL syringe. Adjust the height of the Erlenmeyer flask and the syringe to allow a gravity-fed rate of 1 mL/min.
Note: The correct height depends on the tubing inner diameter (ID). For tubing of 5/32 inches ID, 30 cm are sufficient. - Start bubbling the 4AP-ACSF in the Erlenmeyer flask and in the syringe with a 95% O2/5% CO2 gas mixture.
- Let the 4AP-ACSF flow through the perfusion tubing into a beaker until there is no air inside; then stop the solution flow.
- Connect the heating element at the MEA base to the thermostat. Place the dry MEA chip inside the MEA amplifier and secure the amplifier head. Use a plastic Pasteur pipette to transfer 4AP-ACSF to the inlet and outer reservoir of the recording chamber.
- Secure the heating cannula to a magnetic holder and place its tip inside the recording chamber inlet port; attach the magnetic holder to a magnetic strip on the MEA amplifier head; connect the perfusion tubing to the cannula; connect the cannula to the thermostat.
NOTE: The heating cannula should be covered by beveled polytetrafluoroethylene (PTFE) tubing to reach the recording chamber inlet port and to minimize noise due to the metallic material of the cannula. When the inlet reservoir is adequately filled with 4AP-ACSF, drops falling from the perfusion system should not be visible. - Place the suction needle inside the reservoir and verify that there is negative pressure by submerging the suction needle into the ACSF; check for a constant low-frequency suction noise.
Note: The suction needle should be placed so as to allow the 4AP-ACSF to flow just above the brain slice surface. A vacuum line or a low-noise vacuum pump can be used. - Set the flow regulator to allow a flow-rate of 1 mL/min and start perfusing.
NOTE: Gravity-fed perfusion eliminates the noise that might be caused by peristaltic pumps; if peristaltic pumps are preferred, low-noise models are mandatory. - Once 4AP-ACSF is flowing through the cannula, turn on the thermostat. Set the heating cannula to 37 °C and the MEA base to 32 °C to achieve a temperature of 32 – 34 °C inside the recording chamber.
NOTE: CAUTION! Never heat the cannula without solution in it or it might be irreversibly damaged. Set the temperature of the cannula higher than the recording temperature (i.e., 32 – 34 °C) to account for the intrinsic temperature offset between the set value and actual value at the tip of the heating cannula and within the recording chamber. The flow rate, environment temperature, and volume of the recording chamber all influence the temperature of the recording solution. The settings reported in step 11.9 are optimized for the described protocol and equipment. Always check the actual recording temperature using a thermocouple and adjust the settings as needed. Do not heat the MEA base above 34 °C to avoid overheating the brain slice. - Place the external reference electrode in the recording chamber inlet reservoir.
NOTE: Although MEA chips are equipped with an internal reference electrode, this is covered by the custom recording chamber. Thus, an external reference electrode must be used. A saturated KCl pellet is the most practical, since it is ready to use without need for chlorination.
12. MEA Live Mapping
- Once the 4AP-ACSF level and the recording temperature are stabilized as desired, turn the perfusion and the suction stopcocks to the off position to temporarily stop them.
- Quickly transfer one brain slice onto the MEA recording chamber using an inverted glass Pasteur pipette. Adjust its position on the MEA recording area as needed using a fire-polished curled Pasteur pipette (Figure 1C) or a soft compact small brush. Place the hold-down anchor (Figure 1D) on the brain slice. Restart the perfusion and the suction by turning their stopcocks back to the on position.
NOTE: CRITICAL! The slice should be transferred, and the perfusion restarted within 60 s or the tissue might die. The slice hold-down anchor should be kept in 4AP-ACSF to prevent the brain slice from moving while placing the anchor on the brain slice due to differences in superficial tension. The anchor to secure the brain slice onto the MEA can be custom-made using stainless steel wire and nylon thread (Figure 1D) or obtained from commercial sources. Figure 1E shows the final experimental set-up with the MEA chip connected to the amplifier's head: a brain slice resting on the MEA chip within the recording chamber is held down by the custom anchor. The reference electrode (red circle) and the PTFE tubing covering the heating cannula (red arrow) are positioned in the inlet reservoir, whereas the suction needle (blue arrow) is positioned in the outlet reservoir.
- Quickly transfer one brain slice onto the MEA recording chamber using an inverted glass Pasteur pipette. Adjust its position on the MEA recording area as needed using a fire-polished curled Pasteur pipette (Figure 1C) or a soft compact small brush. Place the hold-down anchor (Figure 1D) on the brain slice. Restart the perfusion and the suction by turning their stopcocks back to the on position.
- Take a picture of the brain slice using a camera mounted on an inverted microscope stage.
- Run the script mapMEA on the computer software to start the GUI to map the electrodes.
NOTE: The custom-made software allows the user to select the electrodes that correspond to specific structures of the brain slice. This step is crucial to activate the correct pathway and suppress ictal activity by using electrical stimulation.
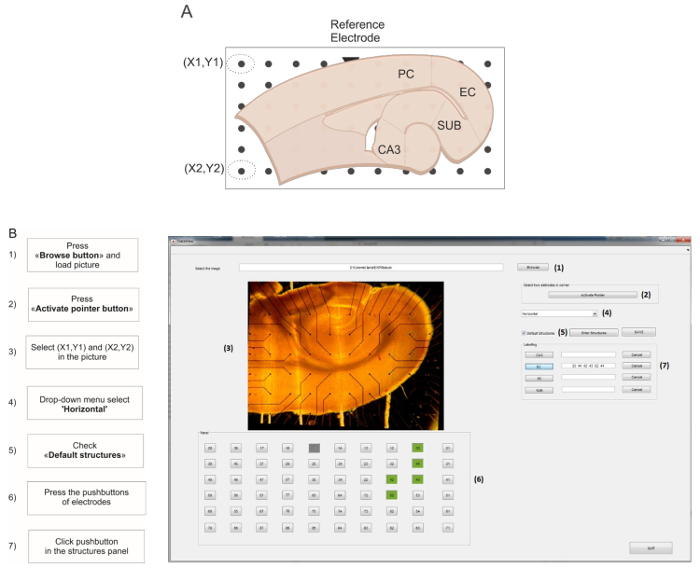
Fig 2: GUI for live MEA electrode mapping. (A) Schematic representation of the combined hippocampus-EC slice positioned on the MEA. The default structures used for this protocol are cornu ammonis 3 (CA3), enthorinal cortex (EC), perirhinal cortex (PC), and subiculum (SUB). The reference electrode is schematized as a triangle marker. Electrodes encircled in dotted lines represent the XY coordinates for image straightening. (B) Screen capture of the GUI during the live MEA mapping. The flow chart on the left of the capture indicates the step-by-step procedure to follow. Please click here to view a larger version of this figure.
- Click the 浏览 button to load the picture of the brain slice. Make sure that the reference electrode appears in the upper row of the left half-side of the MEA (Figure 2A, triangle mark). Click the Activate Pointer button, then select the top and bottom electrodes in the leftmost row of the array to mark the XY coordinates for image straightening and electrode mapping.
- From the slice type drop-down menu select Horizontal. Tick the Default structures checkbox.
NOTE: It is possible to customize the structures by selecting the Enter New Structures button. The default structures for the horizontal brain slice are depicted in Figure 2A. - Using the numbered pushbuttons below the brain slice picture, select the electrodes corresponding to the ROI and click the corresponding pushbutton in the structures panel to assign them (Figure 2B); repeat this step for each ROI.
- Press the Save button: the software generates a result folder named #EXP_LabelledElectrodes containing a table reporting the selected electrodes and ROIs.
13. Recording and Electrical Modulation of the Epileptiform Activity
- Allow the brain slice to stabilize within the recording chamber for 5 – 10 min before recording.
- Turn on the stimulus unit at least 10 min prior to the stimulation protocol to allow self-calibration and stabilization. Start the stimulus control software and verify that stimulator and MEA amplifier are correctly connected as indicated by a green LED in the main panel of the stimulus control software. Please refer to the manuals of the specific instruments and software for additional details, cf. Materials Table.
- Set up the stimulation in bipolar configuration. Select electrode pairs in contact with the pyramidal cell layer of the CA1/proximal subiculum (cf.24,25) among the ones mapped with the script mapMEA (cf. section 12). Use a wire to connect one of the selected electrodes to the negative plug of the stimulator and the other electrode to the positive plug of the same stimulator channel. Use another wire to connect the ground of the stimulator to the ground of the amplifier.
- Start the recording software. To acquire data, press the PLAY button in the main panel of the recording software. Record at least 4 ictal discharges.
NOTE: A sampling frequency of 2 kHz allows acquiring field potentials with fair resolution while minimizing hard disc space usage. A 5-min recording file takes ~80 MB. Higher sampling frequencies may be required, e.g., to record stimulus artifacts or multi-unit activity. To observe field potentials only, use a live low-pass filter at 300 Hz to cut off multi-unit activity. - Determine the stimulus intensity.
- In the stimulus control software, use the main panel to design a square biphasic positive-negative current pulse duration of 100 µs/phase.
NOTE: CAUTION! Direct current stimulation requires a balanced charge pulse to avoid damaging the equipment. - Run a fast input/output (I/O) test to identify the best stimulus intensity. Deliver the stimulation pulse designed at step 13.5.1 at 0.2 Hz or lower by adding an inter-pulse interval of 5 s or longer in the appropriate form of the stimulus control software. In the pulse amplitude tab enter an initial pulse amplitude of 100 µA/phase and increase by 50 – 100 µA steps at each trial until stimulation can reliably evoke interictal-like events in the parahippocampal cortices (check the signals visualized by the recording software).
- In the stimulus control software, use the main panel to design a square biphasic positive-negative current pulse duration of 100 µs/phase.
- Electrical modulation of limbic ictogenesis
- Program the stimulus unit to deliver the stimulation protocol of interest. Use the stimulus amplitude identified during the I/O test.
NOTE: The failure rate of evoked responses should be ≤20%. - After stimulation stops, verify network recovery to pre-stimulus condition by recording at least 4 ictal discharges (as in step 13.4).
Planar MEAs with 6 x 10 layout and 500 µm electrode spacing are the ideal recording device for the experimental protocol described here, since their recording area spans throughout the brain slice in its entirety (Figure 3A, see also17). Although perforated MEAs (pMEAs) would be preferable to improve tissue oxygenation, their recording area is too small (~2 mm diameter). This does not allow the simultaneous visualization of electrical signals generated by the hippocampus and the parahippocampal cortices (data not shown; see18).
The observed 4AP-induced epileptiform pattern (Figure 3A) faithfully reproduces what is typically observed in this in vitro model using conventional field potential recording and comprises three types of activity7,26: (i) ictal-like events (Figure 3B, arrowheads) are robust long-lasting (>20 s, range: 20 – 60 s) events resembling the electrographic features of seizure activity with tonic and clonic components (Figure 3C); they are generated within the parahippocampal cortices every 3 – 5 min and re-enter the hippocampal formation through the dentate gyrus (Figure 3D, arrow); (ii) slow interictal-like discharges (Figure 3B, EC trace, black bar; expanded in Figure 3E) are short (<1 s) population events generated between ictal events ubiquitously within the hippocampus-EC brain slice preparation and occur at a slow pace (range 10 - 30 s); (iii) fastinterictal-like discharges (Figure 3B, CA3 trace, black bar; expanded in Figure 3E) are recurring brief (<1 s) population events generated by the CA3 hippocampal subfield; when the Schaffer Collaterals are preserved, fast CA3-driven interictal events propagate along the hippocampal loop and exert an anti-ictogenic effect24 (data not shown); to prevent ictal activity rundown, for the purpose of the described protocol, CA3 output is disrupted (cf. Figure 3B, E). It needs to be noted that the fast CA3-driven interictal activity recorded using MEAs occurs at a slower pace (~ 0.4 Hz) than what is observed using conventional field potential recording with glass pipettes (~ 1 Hz, range: 0.5 – 2.0 Hz, data not shown).
The successful outcome of electrical neuromodulation in rodent brain slices depends on the activation of specific neuronal pathways27 along with the intrinsic connectivity among the regions included in the preparation24. We have tested slices obtained from adult male Sprague-Dawley rats and CD1 mice and we have found that mouse rather than rat brain slices offer the best trade-off among size, thickness, and intrinsic connectivity. First, thanks to their smaller size, they fit the small recording area of commercially available MEAs better (Figure 4A, left: rat, right: mouse); second, they are more resilient to the disadvantageous condition that is intrinsic to MEA recording (cf. Introduction): although ictal-like discharges generated by these two tissues (Figure 4B) are of similar duration (Figure 4C left), their rate of occurrence is significantly shorter in mouse brain slices (n = 10 slices each species; 2-tailed unpaired t-test, p < 0.001; Figure 4C right). The latter allows speeding-up the experimental protocols, making it possible to test a greater number of brain slices during a single experimental day: for this set of representative results, we could test a similar number of brain slices from the two species (rat: n = 58; mouse: n = 52), but the number of mice (n = 18) was 2-fold smaller than the number of rats (n = 42).
Moreover, mouse brain slices appear to present with denser connections and are therefore more likely to respond better to sustained electrical neuromodulation (Figure 4D). Thus, in this in vitro model of ictogenesis, the viability for electrical stimulation aimed at controlling ictal activity is significantly greater for mouse than for rat brain tissue. This aspect is also reflected by the significantly higher success rate of stimulation experiments performed using mouse versus rat brain slices even when pursuing several sustained stimulation protocols (Figure 4E). In fact, mouse brain tissue appears to better withstand sustained electrical stimulation as suggested by the higher survival rate within the tested experimental time-frame of 4 h (Figure 4F). Thus, the smaller size along with the better preservation of intrinsic connectivity makes mouse brain slices the best candidate to perform MEA recording aimed at studying hippocampal-parahippocampal network interactions and to evaluate electrical neuromodulation protocols that are relevant to TLE.
Periodic pacing delivered in the CA1/subiculum is known to control limbic ictogenesis in the 4AP-treated hippocampus-EC slice preparation24,25,27, with 1 Hz as the most effective frequency27. Thus, it is also useful as a positive control to evaluate the efficacy and efficiency of other neuromodulation policies (see for example25). As mentioned above, electrical pulses delivered directly through the MEA (i.e., without the need of an external stimulating electrode) can evoke population responses in rodent brain tissue (see also28). Sufficient preservation of neuronal network connectivity within the brain slice, accurate positioning of the brain slice onto the MEA, and the appropriate choice of stimulating electrode pairs allow pursuing electrical neuromodulation experiments that effectively control ictogenesis. As shown in Figure 5A, it is possible to reproduce the canonical 1 Hz periodic pacing protocol using planar MEAs both as a recording and as a stimulating device, with the added benefit that the effect of electrical stimulation can be visualized throughout the brain tissue section with a higher number of equally spaced observation points as compared to the conventional field potential recording. Figure 5B shows the quantification of the results obtained for n = 9 brain slices, indicating a significant reduction of the total seizing time (total ictal activity duration/observation time25) during stimulation (one way-ANOVA, F(df): 6.84(2), p <0.01, Fisher's LSD post hoc test, protected). The results are consistent with the literature for external stimulating electrodes24,25.
To compute the total seizing time, the observation time depends on the interval between ictal events, and it also determines the minimum duration of the stimulation protocol required to confidently evaluate the effects of neuromodulation. We find that recording at least 4 ictal discharges (and hence at least 3 intervals between ictal events) is a good trade-off between collected samples and required collection time. In the control condition (i.e., no stimulation), the observation time is the lag between the onset of the first measured ictal event and the termination of the last one. During neuromodulation, the observation time equals the stimulus protocol duration, since the objective of the measurement is to quantify the phenomena occurring throughout the time that the perturbation is introduced in the system. Computing and comparing the total seizing time rather than the duration and interval of ictal events is the most appropriate parameter to avoid type II error. In fact, along with complete suppression of ictal activity, it is also possible that only one ictal event is generated during electrical stimulation as opposed to the several events observed in the control condition. In this case, whereas measuring the interval between events is not possible, measuring ictal duration as an estimator of stimulation efficacy does not represent the actual outcome of neuromodulation (i.e., ictal activity reduction).
Overall, the results presented here indicate that mouse brain slices coupled to MEAs are valuable tools for epilepsy research and to perform reliable neuromodulation studies that are relevant to advance the field of DBS for epilepsy treatment. In addition, the protocol described here improves brain slice viability and allows pursuing experimental sessions for several hours (Figure 6), which may be required, for example, in order to compare the effects of different electrical stimulation paradigms.
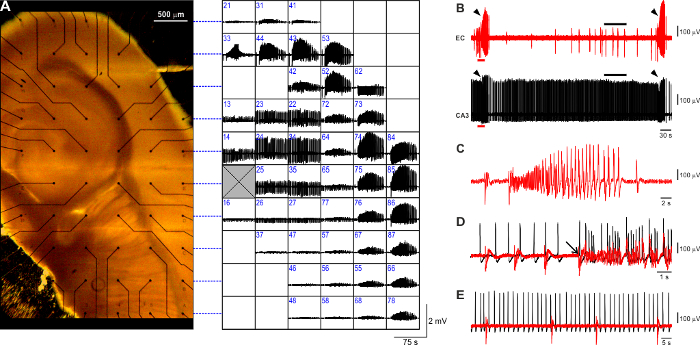
Figure 3: Typical 4AP-induced epileptiform pattern visualized with planar MEAs. (A) Mouse brain slice positioned on a planar 6 x 10 MEA (electrode spacing: 500 µm) and side-by-side overview of the 4AP-induced epileptiform pattern visualized with MEA recording. Each square in the grid accommodates the activity recorded at the corresponding location within the brain slice. Dashed blue lines refer to the electrode rows for more clarity. The recording electrode number is identified in blue at the upper-left corner of each square. The grey crossed square represents the reference electrode. (B) Representative trace segments of EC and CA3 patterns visualized at a faster time-scale. Arrowheads indicate ictal events. In the EC trace it is possible to appreciate the occurrence of slow interictal discharges (black bar), whereas the CA3 subfield generates the typical sustained fastinterictal pattern (black bar). (C) Expanded recording of an ictal discharge showing the typical tonic-clonic pattern. (D) Fast-scale visualization of the pre-ictal-to-ictal transition corresponding to the EC (red) and CA3 (black) trace segments marked by the red bar in (B). The arrow indicates the onset of the ictal discharge. The superimposed traces highlight that the ictal event originates in the EC and subsequently propagates to the CA3 subfield. (E) The expanded view of the interictal periods marked by the black bars in (B) emphasizes the lack of correlation between the slow and fastinterictal patterns generated by the EC (red) and CA3 (black), respectively. Please click here to view a larger version of this figure.
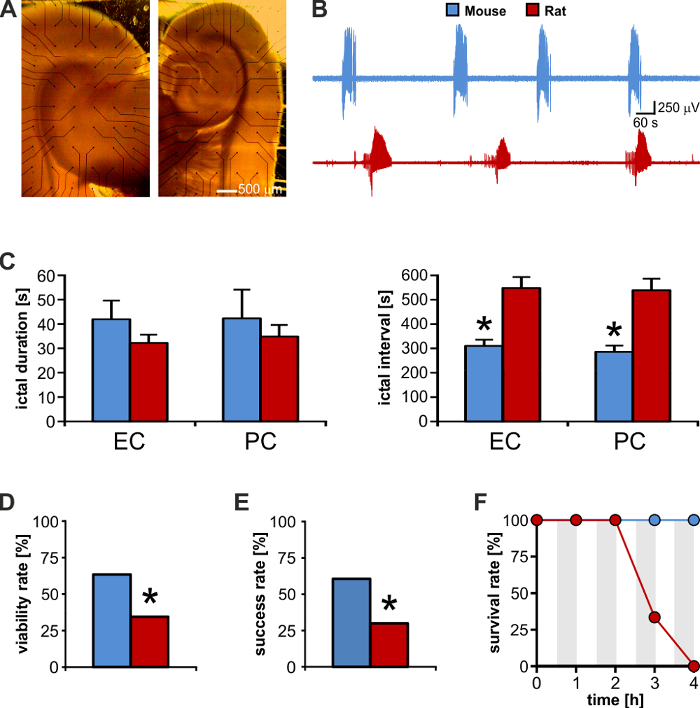
Figure 4: Mouse brain slices offer a higher experimental output than rat brain slices. (A) Brain slice from a rat (left) and a mouse (right) of matched ages. The rat brain slice is much larger than the MEA recording area and its electrical activity cannot be visualized in full. (B) Direct visual comparison of recurrent ictal activity generated by the mouse and rat enthorinal cortex (EC). (C) Mouse and rat brain slices generate ictal discharges of similar duration, but the interval between these events is almost 2-fold in rat brain tissue. * p <0.05. (D) As compared to rats, mice offer a 2-fold higher yield of brain slices viable for electrical stimulation experiments aimed at controlling ictogenesis. Electrical stimulation of the subiculum could evoke population responses in the parahippocampal cortices in only 20 of 58 (34%) rat brain slices, as opposed to 33 of 52 (63%) mouse brain slices (Chi2: 9.22; p = 0.002). * p <0.05. (E) Among viable slices, the success rate in controlling ictal activity is 2-fold with mouse versus rat brain slices (mouse: 20 of 33 brain slices, 60%: rat: 6 of 20 brain slices, 30%; Chi2: 4.67; p = 0.03). * p <0.05. (F) Mouse brain slices can withstand repeated prolonged epochs of sustained electrical stimulation (grey-shaded areas). Upon stimulus withdrawal, mouse brain tissue exhibits full recovery of the epileptiform pattern, whereas rat brain tissue viability falls dramatically at 3 h. Please click here to view a larger version of this figure.
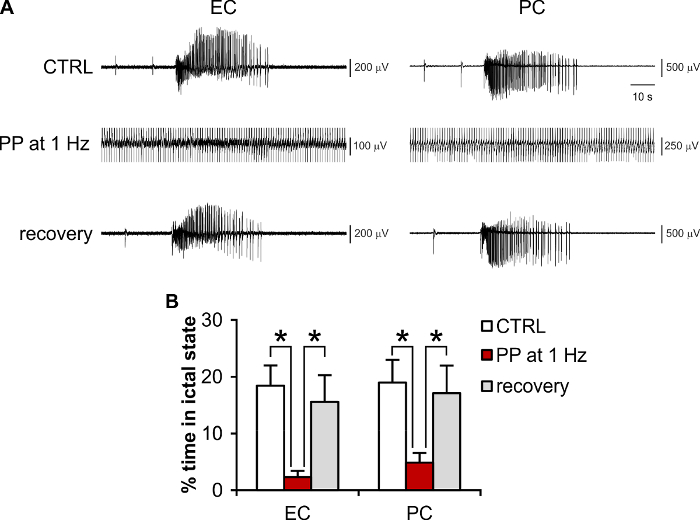
Figure 5: Representative experiment of electrical modulation of ictogenesis using brain slices coupled to MEAs. (A) Recordings of 4AP-induced ictal activity in the EC and PC during the control condition (no stimulation), periodic pacing (PP) at 1 Hz (stimulus artifact is truncated), and during recovery upon stimulus withdrawal. (B) Quantification of the effect of PP at 1 Hz on the total seizing time. * p <0.05. Please click here to view a larger version of this figure.
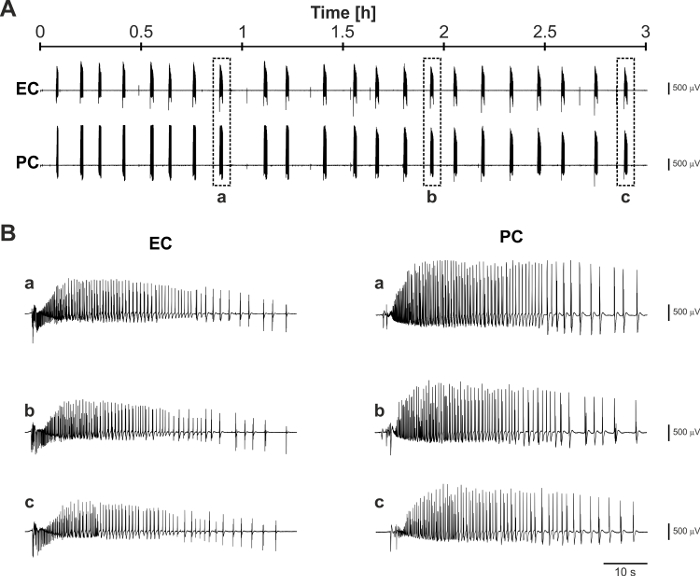
Figure 6: Representative long-term recording of 4AP-induced ictal activity. (A) Trace segments recorded 8 h after the brain slicing procedure and following 2 h of 4AP application. (B) Expanded trace segments corresponding to the boxed ictal discharges identified by the letters a, b, and c in panel (A) show the consistency of generated ictal activity throughout the prolonged observation time-widow. Please click here to view a larger version of this figure.
| Stock A | Chemical | Molecular weight | Concentration (mM) |
| NaCl | 58.44 | 1150 | |
| Solvent: distilled water | KCl | 74.55 | 20 |
| Concentrate: 10x | KH2PO4 | 136.1 | 12.5 |
| Storage temperature: 4 °C | CaCl2*2 H2O | 147.02 | 20 |
| Maximum storage time: 1 wk | D-Glucose | 180.2 | 250 |
| note: filter using a 50 mm membrane filter | |||
| Stock B | Chemical | Molecular weight | Concentration (mM) |
| NaHCO3 | 84.01 | 260 | |
| Solvent: distilled water | |||
| Concentrate: 10x | |||
| Storage temperature: 4 °C | |||
| Maximum storage time: 1 wk | |||
| note: filter using a 50 mm membrane filter | |||
| Stock C | Chemical | Molecular weight | Concentration (mM) |
| KCl | 74.55 | 20 | |
| Solvent: distilled water | KH2PO4 | 136.1 | 12.5 |
| Concentrate: 10x | MgCl2*6 H2O | 203.3 | 50 |
| Storage temperature: 4 °C | MgSO4*6 H2O | 246.47 | 20 |
| Maximum storage time: 2 wks | CaCl2*2 H2O | 147.02 | 5 |
| note: filter using a 50 mm membrane filter | |||
| Stock M | Chemical | Molecular weight | Concentration (mM) |
| MgSO4*6 H2O | 246.47 | 100 | |
| Solvent: distilled water | |||
| Concentrate: 100x | |||
| Storage temperature: 4 °C | |||
| Maximum storage time: 4 wks | |||
| 4AP stock | Chemical | Molecular weight | Concentration (mM) |
| 4-aminopyridine | 94.11 | 250 | |
| Solvent: distilled water | |||
| Concentrate: 1000x | |||
| Storage temperature: 4 °C | |||
| Maximum storage time: 2 wks | |||
| note: vortex for 2 – 3 min or stirr for 30 – 60 min | |||
| Note: CAUTION! 4-AP is toxic and cinvulsant. Use gloves and avoid spreading or spilling. | |||
Table 1: Stock solutions.
| HOLDING ACSF | RECORDING ACSF | CUTTING ACSF | |||||
| Chemical | C [mM] | Chemical | C [mM] | Chemical | C [mM] | ||
| NaCl | 115 | NaCl | 115 | Sucrose | 208 | ||
| KCl | 2 | KCl | 2 | KCl | 2 | ||
| KH2PO4 | 1.25 | KH2PO4 | 1.25 | KH2PO4 | 1.25 | ||
| MgSO4 | 1.3 | MgSO4 | 1 | MgCl2 | 5 | ||
| CaCl2 | 2 | CaCl2 | 2 | MgSO4 | 2 | ||
| D-glucose | 25 | D-glucose | 25 | CaCl2 | 0.5 | ||
| NaHCO3 | 26 | NaHCO3 | 26 | D-glucose | 10 | ||
| L-Ascorbic Acid | 1 | L-Ascorbic Acid | 1 | NaHCO3 | 26 | ||
| L-Ascorbic Acid | 1 | ||||||
| pH | 7.4 | pH | 7.4 | Pyruvic Acid | 3 | ||
| Osmolality | 300 mOsm/Kg | Osmolality | 300 mOsm/Kg | ||||
| pH | 7.4 | ||||||
| Osmolality | 300 mOsm/Kg | ||||||
Table 2: Solutions composition.
| Troubleshooting | |||||
| ISSUE | COUNTERMEASURES | ||||
| Ictal discharges look ‘chunked’ | Decrease the perfusion rate and/or lower the ACSF volume above the brain slice by adjusting the suction needle position. | ||||
| No ictal activity | (1) Verify that CA3-driven fast interictal events do not propagate to the cortex. Use a small scalpel blade (e.g., n.10) or a needle to sever the Schaffer Collaterals if need be, but be careful not to cut the nylon threads of the slice hold-down anchor. A stereo- or upright microscope are best suited, whereas the inverted microscope makes this task very difficult. | ||||
| (2) Check the slice viability: can strong electrical stimulation trigger ictal activity? Wait until about 2 h of 4AP application, then change brain slice if ictal activity does not occur. | |||||
| The brain slice moves when placing the hold-down anchor | (1) Gently remove the slice anchor and reposition the brain slice. | ||||
| (2) Check that the slice anchor is evenly wet of 4AP-ACSF. | |||||
| (3) Remove the 4AP-ACSF from the recording chamber to favor the brain slice adhesion to the MEA. | |||||
| (4) Reposition the slice hold-down anchor. | |||||
| The brain slice floats on the MEA chip after being transferred | (1) Gently aspirate excess ACSF with a Pasteur pipette. | ||||
| (2) The MEA chip might need to be re-coated. | |||||
| Electrical stimulation does not elicit network responses | Increase stimulus intensity, change electrode pairs. | ||||
| Electrical stimulation can elicit population responses in the proximal but not distal cortical areas | The issue is ikely due to poor connectivity or too low stimulus intensity. Electrical stimulation may be ineffective or effective in some cortical areas only. It is recommended to change brain slice. | ||||
| Signal is noisy | (1) Check the reference electrode: noisy baseline can at times be caused by the incomplete fill of the inlet reservoir such that the reference electrodes does not completely deep into the ACSF. | ||||
| (2) Check the overall grounding of equipment. | |||||
| (3) Adjust the suction needle position so as to hear a constant, soft suction sound. Ground the suction needle. | |||||
| (4) Clean the MEA external contacts with ethanol using a cotton swab. | |||||
| (5) The MEA chip may be damaged: change MEA chip and check artifacts and the signal-to-noise ratio. | |||||
Table 3: Troubleshooting.
