This protocol includes 6 major steps following dissection of the ovaries, as outlined in Figure 1. Figure 2, Figure 3, Figure 4 highlight the most novel features of this protocol, which include optimization of tissue clearing for ovaries and whole tissue oocyte quantification using FIJI-ImageJ. Figure 2A shows images of an uncultured 5-day postnatal fixed ovary before (left) and 1 h after (right) adding clearing solution to the ovary. The ovary will begin to become translucent within minutes of being submerged in clearing solution. Once cleared, small ovaries like the one imaged in Figure 2A become difficult to handle without damaging. Therefore, working with cultured explanted ovaries is advantageous because they become attached to the porous surface of the insert membrane on which they are cultured. Attachment of the ovary to the insert membrane allows the experimenter to handle the membrane insert rather than the ovary itself (Figure 2B and Figure 3). Also, ovaries cultured in close proximity will fuse and Figure 2B shows attached fused ovaries before (left) and after clearing (right) on hematoxylin stained samples.
Performing the optical sectioning of cultured ovaries without clearing is possible; however, cells deep within the tissue have a signal that is difficult to differentiate from the background and this lack of clear signal impedes proper oocyte quantification (Figure 3A). In contrast to Figure 3A, Figure 3B is a representative image of a cleared sample in which oocyte quantification throughout the entire organ is possible. Figure 3B demonstrates that whole organ imaging of cleared cultured ovaries can be conducted without significant loss of signal deep within the tissue and images such as the one shown (Figure 3B) can be readily quantified. One approach for quantifying oocytes in samples such as these is by converting the Z-stack series of optical sections into a Maximum Intensity Projection and then further processing the file with an image processing package. Figure 4 and Supplemental Figure 1 highlight the steps used to quantify the number of oocytes in Figure 3B using FIJI-ImageJ. Supplemental Table 1 includes FIJI-ImageJ's particle (oocyte) quantification. Quantification of particles using this method also allows for the analysis of different follicles based on oocyte size, because information on both the number of particles and the corresponding area of each particle is calculated by the software and provided to the user.
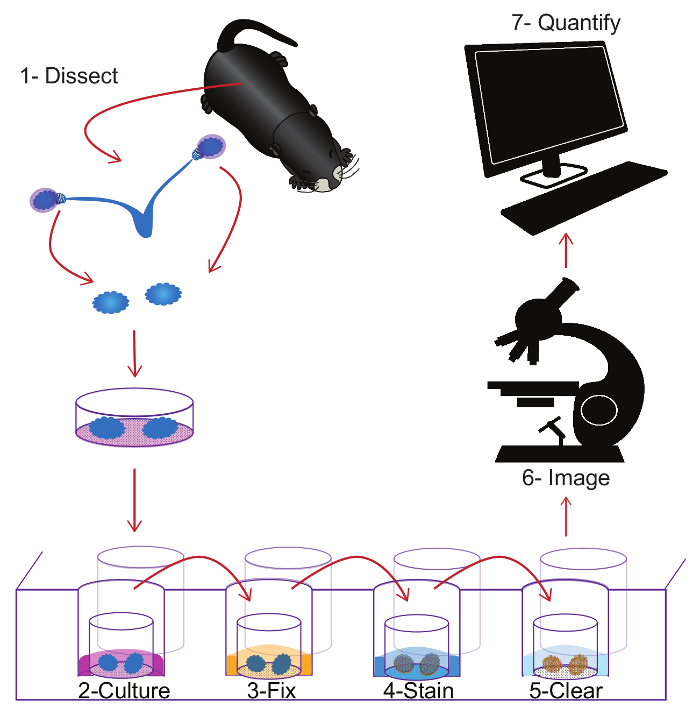
Figure 1: Schematic representation of the entire protocol. Visual summary of the protocol beginning with dissection of the ovary (1), followed by ovary culture (2), tissue fixation with 4%PFA (3), immunostaining using specific antibodies (4), clearing tissue for deep imaging (5), obtaining the optical section using a confocal microscope (6) and ending with oocyte quantification (7). Please click here to view a larger version of this figure.
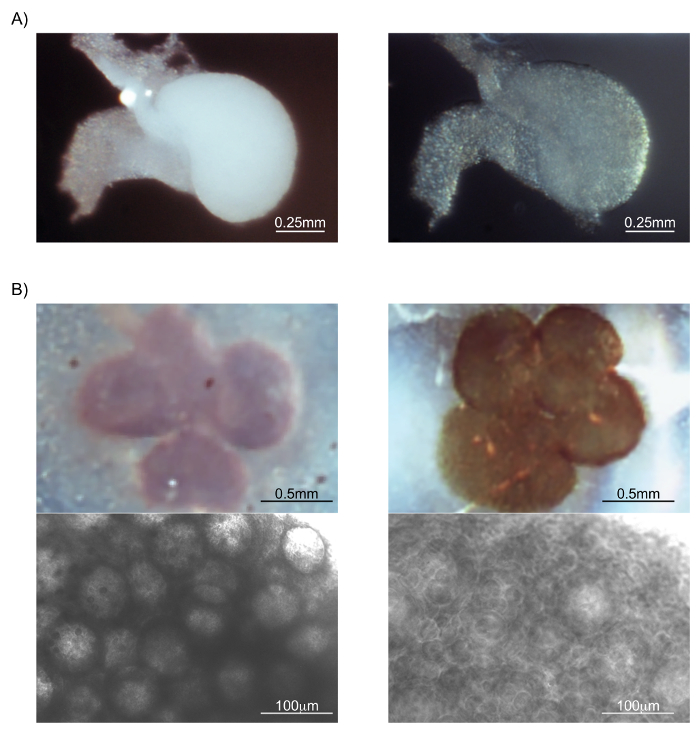
Figure 2: Difference in tissue opacity between un-cleared and cleared ovaries. (A) Ovary from a postnatal day five pup without clearing (left) and after mild clearing of about 1 h (right). (B) Multiple ovaries cultured in close proximity to each other for seven days. Two different magnifications of each sample are shown. Leftmost images are without clearing and rightmost images are with clearing. Ovaries will attach to the membrane of the culture insert and can be better handled if kept attached throughout the protocol. In order to improve the contrast of tissue for imaging using white light, ovaries were submerged in hematoxylin for 5 min after fixation. Please click here to view a larger version of this figure.
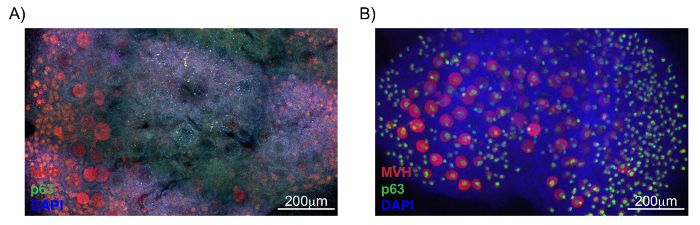
Figure 3: Immunofluorescence imaging of un-cleared and cleared ovaries. Ovaries from postnatal day 5 pups were cultured for 7 days and stained according to the whole mount immunofluorescence staining protocol described. Shown in red are cells labeled with mouse vasa homolog (MVH) to identify germ cells and in green are cells labeled with the nuclear oocyte-specific marker, p63. DAPI, in blue, was used to label all cell nuclei. (A) Immunofluorescence image of ovaries without clearing. (B) Immunofluorescence image of ovaries with clearing. Please click here to view a larger version of this figure.

Figure 4: Visual workflow for how to quantify oocytes using FIJI-ImageJ software. In order to facilitate data analysis, images derived from the confocal planes can be reduced to a maximum intensity projection instead of a 3D image. With the maximum intensity projection, the user can define image threshold parameters in such a way that the target particles become evident. Once the simplified image is obtained, the software is used to count the oocytes. Critically examining images while setting the parameters is crucial. This figure shows an example of how the particle/oocyte threshold can be set. The text above each image highlights the parameter used to obtain that image in FIJI-ImageJ. The software generates a table with the area measurement of the particles (see Supplemental Table 1). Please click here to view a larger version of this figure.
Supplemental Figure 1: Visual workflow for how to quantify oocytes using FIJI-ImageJ software (lower magnification). This figure shows oocyte quantification for two ovaries in close proximity. The steps performed are the same as in Figure 4. 2,436 particles/follicles were counted by the software. Quantification data obtained from the software can be found in Supplemental Table 1. Please click here to download this file.
Supplemental Table 1: Follicle quantification computed by FIJI-ImageJ. This table contains the list of counted follicles and the corresponding area generated from the image in Supplemental Figure 1. The particle size used for quantification was set from 10 µm2-infinity. Please click here to download this file.
