1. Producing BacMam Expression Baculoviruses
- Insert the coding sequence of a desired mammalian bestrophin protein into the pEG BacMam vector18 with a Tobacco Etch Virus (TEV) protease recognition sequence, followed by a GFP-10x His-tag at the C-terminus of the protein.
- Transiently transfect the expression plasmid into adhesive HEK293 cells19,20,21,22,23. Check the expression of the GFP-fusion protein under a fluorescent microscope with 10X or 20X magnification, a 488 nm excitation source, and a 510 nm emission filter (Figure 1A).
Note: Polymer-based transfection is routinely performed with 1 μg of plasmid DNA for a 35 mm dish of HEK293 cells. - Produce the baculoviruses in Sf9 insect cells as previously described16,17.
Note: The passage 3 (P3) virus, which will be used to infect the HEK293-F cells for protein production, can be stored at 4 °C for 1 month. - Determine the titer (infectious units) of the P3 virus by the plaque formation assay16,17.
Note: As the protein of interest is tagged with GFP, a faster method for checking infectious units is to infect Sf9 cells with serial dilutions of the virus and count the number of fluorescent cells after 48 h.
2. Protein Expression
- Maintain the HEK293-F cells in a suspension culture with the HEK293-F expression medium in a 37 °C humidified incubator with 8% CO2. Use disposable culture flasks that are 2-2.5 times bigger than the culture volume and rotate the culture on an orbital platform at 135 rpm.
Note: It is recommended to infect the cells when they are between passages 5 and 30 and to perform these actions under a cell culture hood. - 24 h before viral infection, add 15 μL of Trypan blue to a 15 μL aliquot of cell culture, and check the cell density and viability using a hemocytometer under a microscope. Dilute the cells to 0.6 x 106 cells/mL in 2 x 2 L flasks each, with 500 mL of medium pre-warmed at 37 °C.
Note: Dead cells are stained blue by Trypan blue. The desired cell viability is >95%. - On the day of infection, check the cell density and viability (step 2.2).
Note: A high cell viability of >95% is essential for efficient infection and protein expression. The expected cell density is ~1.0 x 106 cells/mL (grown overnight from 2 x 500 mL of the 0.6 x 106 cells/mL cell culture). The expected total number of cells is ~1.0 x 109. - Add the P3 baculoviruses to the cells at a multiplicity of infection (MOI) of 5. To determine the amount of virus to inoculate, use the following equation:
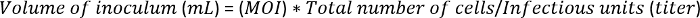
- Shake the cells on the orbital platform at 135 rpm in a humidified 37 °C incubator with 8% CO2 for 24 h.
- Add 10 mM of sodium butyrate to the cell culture and continue incubating for 48 h.
Note: For some proteins, reducing the cell culture temperature to 30 °C after sodium butyrate addition may improve expression and stability. - Under a fluorescence microscope with 10X or 20X magnification, a 488 nm excitation source, and a 510 nm emission filter, check the percentage and brightness of green cells, which directly represent the protein (bestrophin-GFP-10xHis) yield (Figure 1B).
Note: The percentage of green cells is calculated by: 100 x (Green cells/Total cells). Good protein expression level is indicated by strong green fluorescence under the microscope. - Harvest the cells by centrifuging at 1,000 x g in 4 °C for 20 min. Remove the supernatant and re-suspend the cell pellets with phosphate-buffered saline (PBS) to a final volume of ~80 mL. Split the cell suspension into 2 x 50 mL conical tubes and centrifuge at 1,000 x g in 4 °C for 20 min. Store the cell pellets at -80 °C.
3. Protein Purification
- Incubate the conical tubes containing the cell pellets in a stirring water bath at room temperature for 10-15 min so that they thaw. Re-suspend the cells in 2x (w/v) volume of buffer A (Table 1) (e.g., 10 g of cell pellets in 20 mL of buffer) supplemented with proteinase inhibitors (Aprotinin, Leupeptin, Pepstatin A and phenylmethylsulfonyl fluoride) at 0.1-1.0 mM. Pipet up and down extensively to obtain a homogenous cell suspension.
- Lyse the cells using a high-pressure homogenizer. Run the cell suspension through the homogenizer at 7-10 MPa for a total of 3-4 times to achieve complete homogenization. Keep the cell lysate on ice for 2-3 mins between rounds.
- Add detergent [e.g., 2% w/v sol-grade n-Dodecyl-β-D-Maltopyranoside (DDM)] to the cell lysate. Incubate with agitation for 1 h at 20 °C to extract the membrane proteins.
Note: The type of detergent needs to be optimized for each protein target18,24. - Centrifuge at 150,000 x g in an ultracentrifuge at 4 °C for 1 h.
Note: All of the following steps are performed at 4 °C. - Collect the clear cell lysate and flow it through a 5 mL His Trap Ni2+-NTA affinity column pre-equilibrated with buffer A (Table 1).
Note: After centrifugation, the clear cell lysate will be sandwiched between a pellet at the bottom and a cloudy layer on top. Use a 10 mL transfer pipet to carefully collect the clear lysate and then switch to a 1 mL pipet for the last few milliliters of lysate. Avoid the pellet, which can clog the Ni2+ column; however, a small amount of the top layer is fine or can be filtered out with a 1.5 μm filter. - Wash the column with 25 mL of buffer B and then 40 mL of buffer C (Tables 2 and 3, respectively).
Note: This is a good place to take a break, as total purification may take up to 12 h and the protein can remain stable while attached to the column overnight. - Attach the column to a fast protein liquid chromatography (FPLC) system and elute the protein from the column with 13 mL of buffer D (Table 3) with fractionation. Collect the protein-enriched fractions according to the UV absorbance readout.
Note: The FPLC conditions are as follows: a 1.0 mL fraction size, a pre-column pressure alarm set to 0.3 MPa, and a flow rate of 0.5 mL/min. - Measure the eluted protein product concentration on a microvolume spectrophotometer. Read the absorbance at 280 nm.
- (Optional) To remove the GFP-10xHis tag, add the TEV protease at a 1:1 mass ratio and incubate at 4 °C for 30 min.
Note: The tag may or may not affect the function or structure of the purified channel. Calculate the TEV amount from the volume and concentration of the collected protein from steps 3.7-3.8. For instance, if 10 mL of elution product is collected and measured at 0.1 mg/mL, the total protein mass is 10 x 0.1 = 1 mg, and 1 mg of TEV will be used for digestion. - Concentrate the protein with a 15 mL centrifugal (50 or 100 kDa) filter unit by spinning at 4,000 x g in 4 °C to a final volume of 400-500 μL (for a 2 mL sample-loading loop on FPLC).
Note: The centrifuging time for concentration varies depending on the concentration and size of the protein. To avoid over-concentrating, which may cause protein precipitation, spin for 10 min at first, then observe the remaining volume and estimate the time for a subsequent spin (e.g., 2-5 min), and so on, to obtain the final volume. Pipet between spins to disperse the protein, which is pulled to the bottom of the filter. - Transfer the concentrated protein to a new 1.5 mL tube and remove any precipitate or bubbles from the concentrated product. Spin at >12,000 x g for 5-10 min at 4 °C and collect the supernatant in a new 1.5 mL tube.
- Load the final product with a 1 mL syringe and a round-tip needle on a FPLC system for size-exclusion chromatography with a size-exclusion column pre-equilibrated with buffer E (Table 5).
Note: The FPLC conditions are as follows: a 0.5 mL fraction size, a pre-column pressure alarm set to 2.50 MPa, a flow volume set to 30 mL of buffer E (Table 5), and a flow rate set to 0.5 mL/min. - Note that well-behaved proteins run as a single peak (Figure 2A) and collect the protein fraction(s) corresponding to that peak (Figure 2A).
- Concentrate the protein with a 4 mL or 0.5 mL centrifugal filter unit (of the same molecular weight cut-off as in step 3.10) and spin at 4,000 x g at 4 °C to a final concentration of 5-10 μg/μL. Use the spectrophotometer to check the final product concentration (step 3.8).
Note: Centrifugation time varies, as the final product volume can differ between purification trials. Usually, centrifugation takes 10-20 min. It is recommended to pipet between spins to disperse the protein, which is pulled to the bottom of the filter. - Transfer the concentrated protein to a new 1.5 mL tube. Remove any precipitate by centrifuging at >12,000 x g for 5-10 min at 4 °C. Transfer the supernatant to a new 1.5 mL tube. Make 10 μL aliquots for storage at -80 °C and save one 2-5 μL small aliquot to run on a 4-15% gradient SDS-PAGE gel at 9 V/cm (Figure 2B).
- Confirm the identity of the purified protein by mass spectrometry8.
Note: In vitro analysis of the function and structure of the purified protein can be performed through planar lipid bilayer and X-ray crystallography, respectively5,8.
The fluorescence intensity in transiently-transfected adhesive HEK293 cells (Figure 1A) is a good indicator for the projected protein expression level in suspension HEK293-F cells (Figure 1B). If the target protein is not well-expressed or is mis-localized in HEK293 cells after transient transfection, it is recommended to consider modifying the expression construct (e.g., changing the position of the GFP tag or making mutations/truncations on the target protein). Small HEK293-F suspension cultures (e.g., 25 mL cultures) are used to optimize the conditions for protein expression, among which the infection MOI and culturing temperature have been the most important in previous experiences.
A successful purification is indicated by a single main peak at the expected elution volume in size-exclusion chromatography (Figure 2A) and a single dominant band on a denatured SDS-PAGE gel (Figure 2B). If multiple peaks appear in size-exclusion chromatography, it is important to collect each peak and run a native gel for determining which peak contains the functional channel pentamers. It should be noted that between two protein constructs, the expression level, indicated by fluorescence intensity at the time of harvest, does not nesessarily correlate with the final yield, as each protein construct behaves differently during purification. For a well-behaved bestrophin protein, 200-500 µg of the final purified product can typically be obtained from 1 L of HEK293-F suspension cells.
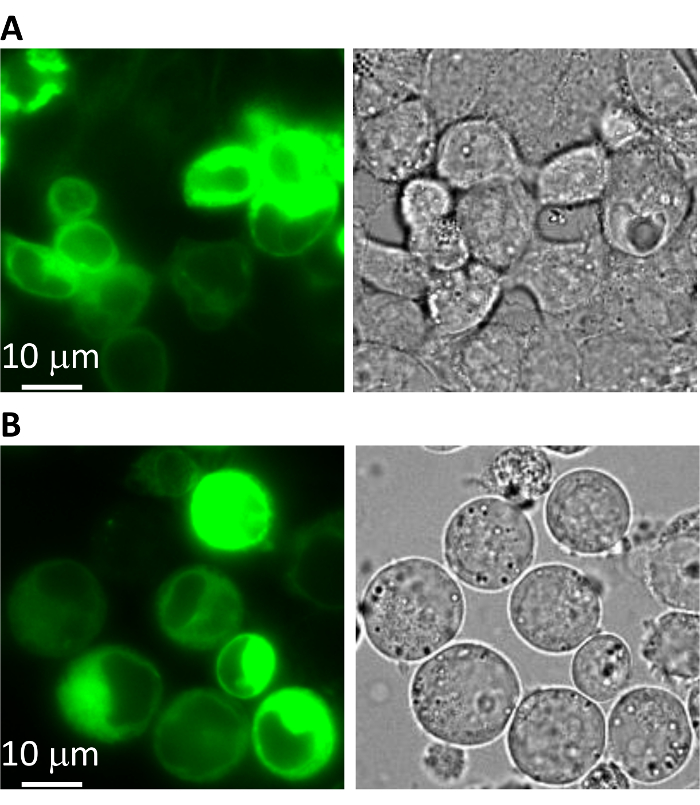
Figure 1: Expression of a mammalian bestrophin protein. Microscopic images of a GFP-tagged mammalian bestrophin expressed in (A) adhesive HEK293 cells by plasmid transfection, and in (B) suspension HEK293F cells by baculovirus infection. Left = green fluorescence; right = bright-field. Please click here to view a larger version of this figure.
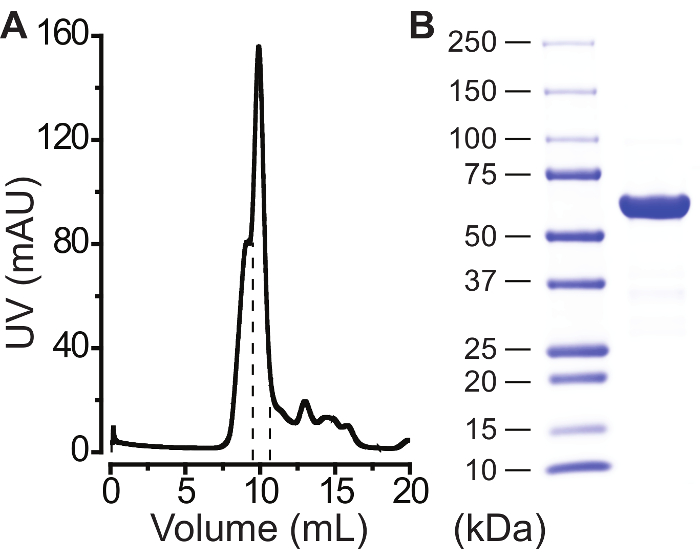
Figure 2: Purification of a mammalian bestrophin protein. (A) Affinity-purified GFP-tagged proteins ran on a size exclusion gel-filtration column as one main peak. Fractions between dashed lines were collected. (B) Final protein product ran on a SDS-PAGE gel with ladders (left column). Please click here to view a larger version of this figure.
| Reagent | MW | Amount per 1 L | Final |
| HEPES | 238.3 | 11.9 g | 50 mM |
| NaCl | 58.44 | 17.529 g | 300 mM |
| Glycerol | 92.09 | 50 mL | 5% (v/v) |
| Imidazole | 68.1 | 1.3616 g | 20 mM |
| MgCl2 | 203 | 0.20331 g | 1 mM |
| tris(2-carboxyethyl)phosphine | 287 | 2.5 mL 200 mM stock | 0.5 mM |
Table 1: Protein Purification Buffer A (Resuspension Buffer)
| Reagent | MW | Amount per 1 L | Final |
| HEPES | 238.3 | 11.9 g | 50 mM |
| NaCl | 58.44 | 17.529 g | 300 mM |
| Glycerol | 92.09 | 50 mL | 5% (v/v) |
| Imidazole | 68.1 | 2.7232 g | 40 mM |
| MgCl2 | 203 | 1.01655 g | 5 mM |
| tris(2-carboxyethyl)phosphine | 287 | 0.5 mL 200 mM stock | 0.1 mM |
| DDM (anagrade) | 510.6 | 0.5 g | 0.05% (w/v) |
Table 2: Protein Purification Buffer B (First Wash Buffer)
| Reagent | MW | Amount per 1 L | Final |
| HEPES | 238.3 | 5.95 g | 25 mM |
| NaCl | 58.44 | 29.2 g | 500 mM |
| Glycerol | 92.09 | 50 mL | 5% (v/v) |
| Imidazole | 68.1 | 5.1 g | 75 mM |
| tris(2-carboxyethyl)phosphine | 287 | 0.5 mL 200 mM stock | 0.1 mM |
| DDM (anagrade) | 510.6 | 0.5 g | 0.05% (w/v) |
Table 3: Protein Purification Buffer C (Second Wash Buffer)
| Reagent | MW | Amount per 1 L | Final |
| HEPES | 238.3 | 7.74 g | 32.5 mM |
| NaCl | 58.44 | 11.686 g | 200 mM |
| Glycerol | 92.09 | 25 mL | 2.5% (v/v) |
| Imidazole | 68.1 | 17.02 g | 250 mM |
| tris(2-carboxyethyl)phosphine | 287 | 0.5 mL 200 mM stock | 0.1 mM |
| DDM (anagrade) | 510.6 | 0.5 g | 0.05% (w/v) |
Table 4: Protein Purification Buffer D (Elution Buffer)
| Reagent | MW | Amount per 1 L | Final |
| HEPES | 238.3 | 9.53 g | 40 mM |
| NaCl | 58.44 | 11.686 g | 200 mM |
| tris(2-carboxyethyl)phosphine | 287 | 0.5 mL 200 mM stock | 0.1 mM |
| DDM (anagrade) | 510.6 | 0.5 g | 0.05% (w/v) |
Table 5: Protein Purification Buffer E (Gel Filtration Buffer)
