| Solution | Composition | |
| Cell culture and transfection | ||
| DMEM1 | DMEM 1x | |
| 1% FBS (v/v) | ||
| 1% GlutaMAX (v/v) | ||
| DMEM10 | DMEM 1x | |
| 10% FBS (v/v) | ||
| 1% GlutaMAX (v/v) | ||
| 150 mM NaCl | 4.380 g NaCl | |
| Up to 500 mL Ultrapure water | ||
| AAV purification and desalting | ||
| 5 M NaCl | 146.1 g NaCl | |
| Up to 500 mL Ultrapure water | ||
| 1 M Tris HCl (pH 8.5) | 12.11 g Tris base | CAUTION |
| Up to 100 mL Ultrapure water | ||
| Add 1 M HCl using a Pasteur pipette to reduce the pH to 8.5 | ||
| Lysis buffer | 15 mL of 5 M NaCl | |
| 25 mL of 1 M Tris HCl (pH 8.5) | CAUTION | |
| Up to 500 mL Ultrapure water | ||
| 10x Phosphate-buffered saline (PBS) | 80 g NaCl | |
| 2 g KCl | CAUTION | |
| 14.4 g Na2HPO4 | ||
| 2.4 g KH2PO4 | ||
| Up to 1 L ddwater | ||
| 1 M MgCl2 | 20.33 g MgCl2-6H2O | |
| Up to 100 mL Ultrapure water | ||
| 1 M KCl | 7.45 g KCl | CAUTION |
| Up to 100 mL Ultrapure water | ||
| 5x PBS Magnesium-Potassium (PBS-MK) stock solution | 250 mL of 10x PBS | |
| 2.5 mL of 1 M MgCl2 | ||
| 6.25 mL of 1 M KCl | CAUTION | |
| Up to 500 mL Ultrapure water H2O | ||
| 15% Iodixanol | 12.5 mL of Optiprep density gradient medium | CAUTION |
| 10 mL of 5 M NaCl | ||
| 10 mL of 5x PBS-MK | ||
| 17.5 mL of Ultrapure water | ||
| 25% Iodixanol | 20.8 mL of Optiprep density gradient medium | CAUTION |
| 10 mL of 5x PBS-MK | ||
| 19.2 mL of Ultrapure water | ||
| 100 µL of phenol red | ||
| 40% Iodixanol | 33.3 mL of Optiprep density gradient medium | CAUTION |
| 10 mL of 5x PBS-MK | ||
| 6.7 mL of Ultrapure water | ||
| 60% Iodixanol | 50 mL of Optiprep density gradient medium | CAUTION |
| 100 µL of phenol red | CAUTION | |
| AAV purity control | ||
| 10x Tris acetate EDTA (TEA) buffer | 44.8 g Tris base | CAUTION |
| 11.4 mL glacial acetic acid (17.4M) | ||
| 3.7 g EDTA | ||
| Up to 1 L Ultrapure water | ||
| Agarose gel | 0.8 g Ultrapure agarose | |
| Up to 100 mL 1x TEA buffer | ||
| Gel buffer | 181.7 g Tris base | CAUTION |
| 1.5 g SDS | CAUTION | |
| Adjust pH to 8.45 with 1 M HCl | CAUTION | |
| Up to 500 mL Ultrapure water | ||
| Cathode buffer 10x | 121.14 g Tris base | CAUTION |
| 179.2 g Tricine | CAUTION | |
| 1% SDS (w/w) | CAUTION | |
| Up to 1 L Ultrapure water | ||
| Anode buffer 10x | 242.3 g Tris base | CAUTION |
| Up to 1 L Ultrapure water | ||
| Adjust pH to 8.9 with 1 M HCl | CAUTION | |
| Sample buffer 5x | For 20 mL: | |
| 605 mg Tris base | CAUTION | |
| 4 g SDS | CAUTION | |
| 10 mg Serva Blue G | ||
| 12 g Glycerol | ||
| Adjust pH to 6.8 with 1 M HCl, aliquot and store at -20 °C | CAUTION | |
| Stacking gel | For 2 gels: | |
| 400 µL acrylamide | CAUTION | |
| 750 µL gel buffer | ||
| 1.85 mL Ultrapure water | ||
| 4 µL TEMED | CAUTION | |
| 20 µL 10% APS (v/v) | CAUTION | |
| Add TEMED and 10% APS immediately before pouring the gel. Use both chemicals under a chemical hood. | ||
| Resolving gel | For 2 gels: | |
| 3.32 mL acrylamide | CAUTION | |
| 3.35 mL gel buffer | ||
| 1.14 mL Ultrapure water | ||
| 2.12 mL 50% glycerol | ||
| 6 µL TEMED | CAUTION | |
| 50 µL 10% APS (w/v) | CAUTION | |
| Add TEMED and 10% APS immediately before pouring the gel. Use both chemicals under a chemical hood. | ||
| Water-saturated butanol | 10 mL n-butanol | CAUTION |
| 1 mL Ultrapure water | ||
| CAUTION: Refer to the Materials Table for guidelines on the use of dangerous chemicals. | ||
Table 1: Composition of the required solutions.
1. Tri-transfection of HEK293T Cells
NOTE: Please refer to Table 1 for the composition of the buffers and solutions used in the protocol.
NOTE: Performance of this section of the protocol takes approximately 4 days.
- Thaw a vial of human embryonic kidney (HEK) 293T cells in a water bath set at 37 °C.
NOTE: Use only cells that have been passaged less than 20x to guarantee optimal transfection efficiency. - Seed HEK293T cells at a density of 2 x 103 to 6 x 103 cells/cm2 in DMEM10 in 15 cm diameter cell culture dishes.
- Grow the cells to 70–80% confluence in a standard incubator set at 37 °C, with 95% humidity and 5% CO2.
NOTE: The production of one batch of AAV vectors using this protocol requires 18 cell culture dishes (15 cm diameter). A cell confluence of 70% – 80% corresponds to 6 x 103 to 7 x 103 cells/cm2, maintained in 17–20 mL of DMEM10 culture medium. - Prepare polyethylenimine (PEI)/DNA mix at a concentration ratio of 1/3.5 (w/w).
- Prepare the DNA mix for 18 cell culture dishes in one 50 mL conical tube by mixing 360 µg of pΔF6, 180 µg of pCapsid, and 180 µg of pTransgene in 18 mL of 150 mM NaCl.
- Distribute the DNA mix over three 50 mL conical tubes (6 mL of DNA mix per conical tube).
- Prepare the PEI mix for six cell culture dishes in a new 50 mL conical tube by mixing 840 µg of PEI (1 µg/µL) in 6 mL of 150 mM NaCl.
- Prepare the PEI/DNA mix by adding 6 mL of the PEI mix (prepared in step 1.4.3), drop by drop, to one of the conical tubes containing the DNA mix (prepared in step 1.4.2) and incubate for 20 min at room temperature.
NOTE: After 20 min of incubation, the PEI/DNA mix will become slightly turbid.
- Take six cell culture dishes out of the incubator and completely aspirate the medium from each culture dish in a laminar flow hood. Remove the traces of the medium by rinsing the dishes with 5 mL of prewarmed Dulbecco’s phosphate-buffered saline (DPBS).
- Ensure the distribution of DPBS over the entire surface by gently tilting the dish.
- Gently aspirate the DPBS and add 12 mL of DMEM1 to each dish.
NOTE: Avoid detaching the cells by adding the medium slowly, from a pipette placed at the edge of the dish. - Mix the PEI/DNA by pipetting up and down 3x – 5x. Add 2 mL of the PEI/DNA mix to each of the six cell culture dishes in a drop-by-drop fashion, carefully distributing it over the entire surface. Once the mix is added to each dish, place the dishes back in the incubator. Repeat steps 1.4.3– 1.8 for the remaining culture dishes.
- Incubate the transfected cells for 5 h at 37 °C, with 95% humidity and 5% CO2.
- Add an additional 12 mL of DMEM10 to each culture dish without removing the pre-existing medium (total medium volume = 25 mL).
- Incubate the transfected cells up to 72 h post-transfection at 37 °C, with 95% humidity and 5% CO2.
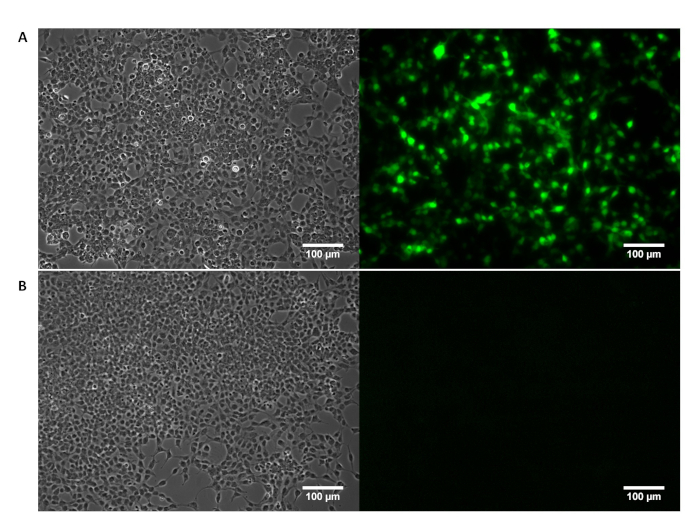
Supplementary Figure 1: HEK 293T cell morphology visualized by phase contrast microscopy (left) with confirmation of GFP expression visualized by fluorescence imaging (right). (A) Successful transfection of HEK293T cells with a GFP-encoding pTransgene is confirmed by fluorescence imaging. (B) HEK293T cells treated solely with transfection reagents show no GFP expression. Please click here to view a larger version of this figure.
- Harvest the medium and the cells 72 h post-transfection. Use a cell scraper to carefully detach the cells from the culture dish. Collect the medium and the cells in a 50 mL conical tube kept on ice.
NOTE: The contents of two cell culture dishes can be collected into a single 50 mL conical tube. At the end of this step, each of the nine tubes will contain approximately 50 mL of medium. - Centrifuge the conical tubes at 420 x g for 10 min at 4 °C with the acceleration and deceleration of the centrifuge set to maximum.
- Carefully discard the supernatant from each tube. Do not use pipettes to prevent the loss of material. Instead, gently pour the supernatant into a waste disposal container and place the tubes containing the cell pellets back on the ice.
- Resuspend each cell pellet in 2 mL of lysis buffer directly in the 50 mL conical tube by pipetting up and down 5–10x. Do not vortex. Pool the lysates from three tubes together.
NOTE: At this point, there will be three 50 mL conical tubes, each one containing 6 mL of the resuspended cells in lysis buffer.
2. AAV Vector Purification
NOTE: Performance of this protocol section takes approximately 1 day. Perform the following steps simultaneously on each of the three 50 mL conical tubes containing the cells resuspended in lysis buffer (see the previous note).
- Freeze and thaw the resuspended cells 3x to lyse them and release the AAV particles. Perform the freezing step by placing the tubes in a bucket containing dry ice mixed with ethanol. Perform thawing by immediately placing the cells in a water bath set at 37 °C.
- After the third thawing step, centrifuge at 1,167 x g for 15 min at 4 °C.
NOTE: A noticeable pellet composed of cell debris will be formed. When handling the tubes, avoid sudden movements as these can cause the detachment of the pellet into the supernatant, which will compromise the purity of the final vector. - Carefully transfer the supernatants to clean 50 mL conical tubes and then add nuclease to each tube, to a final concentration of 50 U/mL supernatant.
- Incubate for 30 min at 37 °C. Swirl the 50 mL conical tubes by hand every 10 min to ensure that the nuclease is thoroughly mixed with the supernatant.
- Clarify the supernatant by centrifugation at 13,490 x g for 20 min at 4 °C.
- Attach a 0.45 µm filter to a 10 mL syringe and place it on top of a clean 50 mL conical tube. Carefully remove the plunger and fill the syringe with the supernatant from step 2.5.
- Use the plunger to force the lysate through the filter. Use a new filter and syringe for each tube of supernatant obtained in step 2.5.
NOTE: The obtained fraction is known as ‘crude lysate’. - Prepare each of the 15%, 25%, 40%, and 60% iodixanol fractions in four separate 50 mL conical tubes, according to the instructions in Table 1.
- Prepare the iodixanol gradients in three ultracentrifugation tubes using the following order of iodixanol fractions: 8 mL of 15% iodixanol, 5.5 mL of 25% iodixanol, 5 mL of 40% iodixanol, and 4.5 mL of 60% iodixanol.
CAUTION: Iodixanol can cause irritation to eyes, skin, and the gastrointestinal and respiratory tracts. When handling iodixanol gradients, wear gloves and work under a laminar flow hood.- Pipette 8 mL of 15% iodixanol into each ultracentrifugation tube.
- Pipette 5.5 mL of 25% iodixanol solution into a clean 50 mL conical tube (Figure 1A).
- Use a non-graduated Pasteur pipette (as the neck of the ultracentrifugation tube is too narrow for conventional graduated pipettes) to carefully layer 5.5 mL of the 25% iodixanol solution below the 15% iodixanol solution (Figure 1B).
NOTE: This can be successfully achieved by adding the iodixanol in three steps since the Pasteur pipette can only hold around 2 mL. - Add the 40% and 60% iodixanol solutions as described in step 2.9.3. Do not disturb the different iodixanol interfaces during layering.
NOTE: The proper preparation of the different iodixanol fractions can be ensured by visual confirmation thanks to the phenol red added to the iodixanol fractions (see the instructions in Table 1). Since each fraction has a specific density, they will not intermix during the layering step, if it is performed properly (check Figure 1C).
- Layer the crude lysate on top of the 15% iodixanol gradient with a Pasteur pipette. Proceed drop by drop to avoid disturbing the interface between the crude lysate and the iodixanol solution.
- Fill the ultracentrifugation tube up with lysis buffer until the meniscus reaches the base of the tube neck to ensure the tube does not collapse under the very high forces generated during the ultracentrifugation (Figure 1C).
- Close the ultracentrifugation tubes using appropriate lids. Use a digital scale to make sure all three ultracentrifugation tubes have the same weight. Adjust the weight, if necessary, by adding more lysis buffer on top of the ‘crude lysate’.
NOTE: The weight difference between the ultracentrifugation tubes must be below 0.1 g to ensure the safe operation of the ultracentrifuge. Ultracentrifuges are potentially dangerous pieces of equipment and should only be used by properly trained personnel. - Centrifuge the tubes at 301,580 x g, using a fixed-angle titanium rotor, for 1 h and 40 min at 12 °C, using maximum speed of acceleration and deceleration.
- Carefully insert a stainless-steel blunt needle at the 40% and 60% iodixanol interface.
- Attach the needle to a 5 mL syringe (Figure 1E).
- Aspirate only the clear fraction, containing the vector particles.
NOTE: The total volume collected is approximately 2.5–3 mL. Avoid the collection of material from the 25–40% interface, since this will increase the level of contamination in the final vector batch, due to the presence of non-desirable proteins. - Process the collected fraction (desalting and concentration) or store it overnight at 4 °C.

Figure 1: Setup for iodixanol gradient purification and subsequent vector collection. (A) Before pipetting the different iodixanol gradients into the ultracentrifugation tube, pipette an adequate volume of each iodixanol solution into a separate conical tube. (B) Then use a Pasteur pipette to sequentially transfer each iodixanol solution to the ultracentrifugation tube: layers of an increasingly high iodixanol concentration should be added at the bottom of the tube underneath the previous layer(s). (C) Layer the crude vector lysate on top once the gradient has been prepared. This vector collection system does not use sharp needles, which present a risk of 'needlestick' injuries. (D) A stainless-steel 316-syringe needle is inserted through the iodixanol gradient up to the 40%/60% iodixanol interface. (E) Vector particles are found in the 40% iodixanol phase and are collected. Please click here to view a larger version of this figure.
3. Desalting and Concentration of the AAV Vector
NOTE: Performance of this section of the protocol takes approximately 2 h.
- Prerinse the filter membrane of the centrifugal filter by adding 5 mL of 1x PBS-MK and centrifuge for 5 min at 4,000 x g.
- Add 5 mL of 1x PBS-MK to the collected AAV vector fraction, mix and then add the total volume to the filter. Upon addition of 1x PBS-MK, turbidity is observed. Centrifuge at 4,000 x g until the volume present on the cone-shaped filter has been reduced to 1 mL. Discard the flow-through accumulated in the outer collection tube.
- Add 13 mL of 1x PBS-MK to the cone-shaped filter and repeat the centrifugation step as many times as necessary (minimum 3x), until there is no more turbidity observed when fresh 1x PBS-MK is added.
- In the final wash step, add 13 mL of PBS + 0.01% (v/v) non-ionic surfactant and centrifuge at 4,000 x g until the volume is reduced to 300–350 µL.
- Collect the remaining fraction present on the filter by pipetting the whole volume into a sterile skirted microcentrifuge tube.
NOTE: This fraction contains the desalted and concentrated AAV vector. In the following steps of this protocol, this fraction is referred to as the ‘primary’ fraction. Make sure to handle the tube under the hood and store it at 4 °C. - Rinse the filter with 200 µL of 1x PBS-MK to collect the residual vector particles retained on the filter. Collect in a sterile microcentrifuge tube.
NOTE: This is a diluted vector stock, which may be suitable for some applications requiring lower vector titers. In future steps, this fraction is referred to as the ‘secondary’ fraction. - For short-term storage, store the vector fraction(s) at 4 °C (less than 2 weeks). Aliquot and store the vector at ‑20 °C when long-term storage is required.
- Perform quality control as described in section 4.
4. Titration of the Vector by Quantitative Polymerase Chain Reaction
NOTE: Performance of this section of the protocol takes approximately 3 h.
- Standard curve
- Linearize the transgene-expressing plasmid (pTransgene) used for the transfection of HEK-293T cells in section 1.
- Prepare the restriction digest mix in a 0.5 mL microcentrifuge tube (refer to Table 2 for the composition of the restriction digest mix).
NOTE: Adjust the composition of the restriction digest mix according to the enzyme used and the related guidelines from the manufacturer. - Incubate the restriction digest mix for 1 h at 37 °C.
- Check the efficiency of the restriction digestion by running the linearized plasmid on a 0.8% (w/v) agarose gel at 100 V for 1 h. Complete digestion of the plasmid should produce a single fragment of defined size.
- Purify the linear plasmid DNA using a PCR purification kit, following the manufacturer’s instructions24, and measure the DNA concentration with a spectrophotometer by measuring the absorption at 260 nm. After the titration, store the remaining aliquot of the linear plasmid at ‑20 °C for further use.
- Calculate the molecules (i.e. DNA copies) of the plasmid stock per microliter as follows.
- First, calculate the molecular weight of the plasmid. Assuming that the average weight of a DNA base pair (bp) is 650 Daltons (Da) and that one mole of a bp weighs 650 g, the molecular weight of any double-stranded DNA template can be estimated by taking the product of its length (in bp) and weight per base pair.
Plasmid molecular weight [g/mol] = plasmid size [bp] x 650 [Da/bp] - Next, calculate the number of moles of plasmid per microliter.
Moles of plasmid per microliter = plasmid concentration [g/µL] / plasmid molecular weight [g/mol]
NOTE: The inverse of the molecular weight is the number of moles of plasmid present in 1 g of the material. - Then, calculate the number of plasmid molecules per microliter, using Avogadro’s number (6.022 x 1023 molecules/mole).
Molecules of plasmid per microliter = moles of plasmid per microliter [moles/µL] x Avogadro’s number [molecules/mole] - Finally, dilute the plasmid stock (molecules/µL) to obtain a 100 µL solution with the desired concentration of 1 x 109 molecules (vector genomes (vg) per microliter).
Plasmid stock (100 µL) = (desired concentration [molecules/µL] x 100 µL) / plasmid molecules [molecules/µL]
- First, calculate the molecular weight of the plasmid. Assuming that the average weight of a DNA base pair (bp) is 650 Daltons (Da) and that one mole of a bp weighs 650 g, the molecular weight of any double-stranded DNA template can be estimated by taking the product of its length (in bp) and weight per base pair.
- Make serial dilutions of the plasmid stock (1 x 109 vg/µL) in triplicates:
10 µL of 1 x 109 vg/µL plasmid stock + 90 µL of H2O = 1 x 108 vg/µL solution
10 µL of 1 x 108 vg/µL dilution + 90 µL of H2O = 1 x 107 vg/µL solution
10 µL of 1 x 107 vg/µL dilution + 90 µL of H2O = 1 x 106 vg/µL solution
10 µL of 1 x 106 vg/µL dilution + 90 µL of H2O = 1 x 105 vg/µL solution
Continue to obtain a 1×101 vg/µL solution. - Keep the serial dilutions of the standard plasmid stock on ice until loading them on the qPCR plate (section 4.3).
| Component | Amount |
| 10x Restriction enzyme buffer | 5 µL |
| Restriction enzyme | 2,5 µL |
| Plasmid | 5 µg |
| H2O | up to 50 µL |
Table 2: Restriction digest mix composition.
Table 3: Stock plasmid volume calculator. Please click here to download this table.
- DNA extraction from the AAV vector
- Mix 2 µL of the AAV vector stock (the primary fraction from step 3.5) with 198 µL of DNase I buffer (1x) in strip tubes (PCR tubes) and add 2 µL of DNase I.
NOTE: DNase I will degrade any genetic material that is not contained inside a vector capsid (which would distort the qPCR results). This solution is referred to as dilution ‘dil1 x 10-2’. - Incubate for 30 min at 37 °C, followed by 10 min at 95 °C.
NOTE: The protocol can be stopped at this point and the material can be stored indefinitely at 4 °C, to avoid product deterioration. - Add 2 µL of proteinase K to the dil1 x 10-2 solution (step 4.2.1) and incubate for 60 min at 50 °C, followed by 20 min at 95 °C.
NOTE: This step will disassemble the AAV vector capsid and release the AAV vector genome into the solution. Add proteinase K in excess as the protein (capsid) content of the sample is not known. Note, it is essential to ensure that all proteinase K activity is removed by denaturation, prior to qPCR, to avoid issues with (partial) polymerase degradation influencing the final result. - Prepare 1:10 serial dilutions of the proteinase K treated dil1 x 10-2 solution (step 4.2.3) in 1.5 mL microcentrifuge tubes as follows:
10 µL of dil1 x 10-2 + 90 µL of H2O = dil1 x 10-3 dilution
10 µL of dil1 x 10-3 + 90 µL of H2O = dil1 x 10-4 dilution
10 µL of dil1 x 10-4 + 90 µL of H2O = dil1 x 10-5 dilution - Keep the serial dilutions of DNA extracted from the vector on ice until loading them on the qPCR plate (section 4.3).
- Mix 2 µL of the AAV vector stock (the primary fraction from step 3.5) with 198 µL of DNase I buffer (1x) in strip tubes (PCR tubes) and add 2 µL of DNase I.
- Titration by SYBR Green detection-based qPCR
- Prepare the qPCR master mix in a 1.5 mL microcentrifuge tube, for both the sample and the standards. Use 10 µL of SYBR Green Master Mix, 1 µL of forward primer (10 µM stock), 1 µL of reverse primer (10 µM stock), and 3 µL of H2O per reaction. See the protocol in Table 4 for primer sequences.
- Pipette the qPCR master mix up and down, but do not vortex.
- Add 15 µL of the qPCR master mix, followed by either 5 µL of the standard curve prepared in section 4.1 or the DNA extracted from the AAV vector in section 4.2, into each well. Include three wells containing only the qPCR master mix, as a negative control. Refer to Figure 2 for the plate layout.
- Seal the plate with a sealing film and briefly centrifuge the qPCR plate at 1,500 x g for 30 s at 4 °C.
- Run the qPCR reaction on a plate-based real-time PCR amplification and detection instrument, using the conditions suggested in Table 5.
| Primer name | Sequence |
| Forward primer | 5’-CCCACTTGGCAGTACATCAA-3’ |
| Reverse primer | 5’-GCCAAGTAGGAAAGTCCCAT-3’ |
Table 4: Primer sequences designed against the CBA promoter.
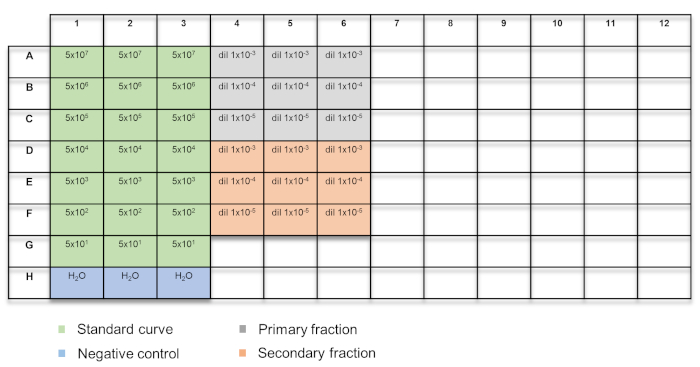
Figure 2: Plate layout for qPCR-based vector titration. The samples are color-coded: green = standard curve; blue = H2O control; grey = primary fraction; orange = secondary fraction. Please click here to view a larger version of this figure.
| Step | Time | Temperature | Cycles | Aim |
| Pre-incubation | 5 min | 95 °C | x1 | DNA denaturation and polymerase activation (hot-start reaction). |
| Amplification | 10 min | 95 °C | x1 | Amplification of the DNA. Settings may be optimized if alternative primers with different annealing temperature are used. |
| 10 s | 95 °C | x40 | ||
| 40 s | 60 °C | |||
| 1 s | 72 °C | |||
| Cooling | 10 s | 40 °C | x1 | Plate cooling. End of the PCR. |
Table 5: Thermal cycling protocol for SYBR green-based qPCR titration.
- Data analysis to determine the AAV vector titer
- Fill the spreadsheet data cells (Table 6A) with the Ct values obtained for the different dilutions of the standard sample prepared in section 4.1 to generate a standard curve.
NOTE: The equation of the standard curve will be shown (y = a ln(x) + b) together with the R2 efficiency (Table 6B). A qPCR must have an efficiency close to 100% and R2 close to 1.0 (≥ 0.96). - Use the calculated values of a and b to fill in the corresponding data cells in the spreadsheet (Table 6C).
- Complete the spreadsheet with the Ct values obtained for the different dilutions of the AAV sample prepared in section 4.2.
- Calculate the average AAV vector titer in vector genomes per microliter of the 'primary fraction' by using the following formula:
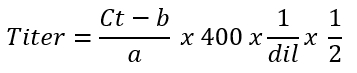
NOTE: The factor 400 is used for single stranded genomes, while sc genomes use a multiplication factor of 20025. In fact, as DNA quantities double during each qPCR cycle, sc DNA is detected 2x in comparison to an ss genome. The aim of the titration is to calculate the concentration in terms of vector genomes per microliter. A correction is necessary for running the qPCR when using 2 µL of starting material (step 4.2.1). dil represents the dilution factors from step 4.2.4. The titer of the 'secondary' AAV vector fraction (step 3.6) can be calculated simultaneously.
- Fill the spreadsheet data cells (Table 6A) with the Ct values obtained for the different dilutions of the standard sample prepared in section 4.1 to generate a standard curve.
Table 6: Template for qPCR data analysis. Please click here to download this file.
5. Purity Control by SDS-PAGE and Silver Staining
NOTE: Performance of this section of the protocol takes approximately 5 h.
- Use 70% (v/v) ethanol to thoroughly clean the glass plates used for casting the gels.
- Assemble the gel casting system. Ensure that the bottoms of the glass plates are not chipped, to prevent leakage of the acrylamide mixture when casting the gel.
- Prepare the stacking and the resolving gel solutions in two separate 50 mL conical tubes. Omit the tetramethylethylenediamine (TEMED) and the 10% (w/v) ammonium persulfate (APS), as they are responsible for polymerization of the gel. Add them immediately before casting the gel.
NOTE: The final percentage of acrylamide in the gel influences the separation profile of the proteins: typically, a 10% (v/v) final concentration of acrylamide will be sufficient to test the purity of an AAV vector preparation. - Mix gently by swirling the tube containing the gel components. Do not vortex, since excessive oxygenation can impair polymerization.
- Add TEMED and 10% (w/v) APS to the resolving gel solution. Mix and then pour into the gel holder until it reaches 1-2 cm below the top of the small glass plate.
- Place a layer of water-saturated butanol on the top of the acrylamide mixture. This will ensure the formation of a flat surface during polymerization. Do not leave the gel under alcohol for longer than 30 min as this will dehydrate the gel and impair its function.
CAUTION: Butanol is hazardous in case of skin contact. It also presents a fire risk. Handle with care. - Wait for the gel to polymerize and then pour off the butanol. Tip: Check to see whether the excess gel mix in the tube has solidified. Polymerization occurs in less than 20 min. Wash the surface of the gel with H2O and then dry it using paper towels, taking care not to disturb the surface of the gel.
- Add TEMED and 10% (w/v) APS to the stacking gel and pour the gel into the gel holder on top of the separating gel.
- Place the provided comb in the gel. Perform this action with one steady movement to avoid the formation of air bubbles inside the wells.
- Wait for at least 20 min for the gel to polymerize. Tip: Check if the excess gel mix in the conical tube has solidified.
- Prepare the two mixes (Table 7) for both the primary and the secondary AAV vector fractions (steps 3.5 and 3.6, respectively).
| High Amount | Low Amount | |
| AAV vector stock | 5 µL | 1 µL |
| 5x Sample buffer | 3 µL | 3 µL |
| H2O | 7 µL | 11 µL |
Table 7: Composition of the sample mixes required for silver staining.
- Fully denature the sample mixes by heating them for 5 min at 95 °C. Assemble the electrophoresis tank, paying attention to the electrode orientation.
- Fill the tank with 1x cathode buffer on the inside of the electrode assembly and 1x anode buffer on the outside.
NOTE: Anode buffer can be recycled from run-to-run. Fresh cathode buffer must always be used. - Remove the comb from the gel and clean the wells of the gel with 1x cathode buffer.
- Load 1 µL of protein ladder in a well. Load the sample mixes in different wells on the same gel (Figure 3). Run the gel at 50 V until the samples enter the resolving gel. Then increase the voltage to 100 V until the dye front reaches the lower limit of the gel.
- Extract the gel carefully from the glass plates and use the silver staining kit (according to the manufacturer’s instructions26) to visualize the viral proteins (VP1, VP2, and VP3 subunits) that comprise the AAV capsid, as well as to check for possible protein contamination (Figure 3).

Figure 3: Evaluation of vector purity using SDS-PAGE and silver staining. Using a Tricine-SDS gel, 5 µL of various vector preparations were separated. Proteins were subsequently detected by silver staining. Vectors are considered pure when VP1 (82 kDa), VP2 (67 kDa), and VP3 (60 kDa) are visible in a 1:1:10 ratio (lane 1), without excessive background (lane 2) or non-specific bands (lane 3). Please click here to view a larger version of this figure.
AAV9 was considered, until recently, to be the most effective AAV vector serotype in crossing the BBB and transducing cells of the CNS, following peripheral administration. A significant advance in capsid design was achieved when Deverman et al. reported the use of a capsid selection method called Cre recombination-based AAV-targeted evolution (CREATE)17. Using this method, they identified a new capsid, named PHP.B, which they reported as able to transduce the majority of astrocytes and neurons in multiple CNS regions, following systemic injection17. At this point, it should be noted that even though PHP.B provides positive results in C57/Bl6 mice (which was the strain used in the initial isolation experiments), preliminary reports suggest its efficiency may vary in a strain-dependent manner. Further experiments will, no doubt, shed more light on this issue31.
However, despite these issues, PHP.B offers exciting possibilities for noninvasive gene manipulation in the CNS of mice, including proof-of-concept gene therapy experiments in disease models. As such, we chose to evaluate the efficiency of transgene expression using PHP.B versus AAV9, which has been the 'gold-standard' vector for CNS transduction following peripheral administration since 20092. To perform a direct comparison of both serotypes, under optimal conditions for transgene expression, we used an sc genome configuration32. Both vectors carried the transgene for green fluorescent protein (GFP) under the control of the ubiquitous chicken β-actin (CBA) promoter. Female C57/Bl6 mice at postnatal day 42 (approximately 20 g in weight) received a dose of 1 x 1012 vg per mouse of either scAAV2/PHP.B-CBA-GFP or scAAV2/9-CBA-GFP. Vector administration was performed via tail vein injection. The experiments were approved by the Ethical Committee of the KU Leuven.
Three weeks postinjection, the mice underwent transcardial perfusion with ice-cold PBS, followed by 4% (w/v) ice-cold paraformaldehyde (PFA). Their brains were harvested and underwent further postfixation by an overnight incubation in the same fixative, before transferring to 0.01% (w/v) Na-azide/PBS for storage until further analysis. Afterward, the brains were sectioned using a vibrating microtome, and immunohistochemistry was performed on 50 µm thick sections.
To evaluate the levels of transgene expression, sections were stained with primary antibodies against GFP (rabbit anti-GFP), with detection using secondary antibodies conjugated to a fluorescent dye (anti-rabbit Alexa Fluor 488) (Figure 4A, B). Fluorescence intensity measurements (in arbitrary units [au]) confirmed a significant increase in GFP expression when an sc genome and the PHP.B capsid were used relative to AAV9. Increases in GFP were observed in the cerebrum (2105 ± 161 vs. 1441 ± 99 au; p = 0.0032), the cerebellum (2601 ± 196 vs. 1737 ± 135 au; p = 0.0032), and the brainstem (3082 ± 319 vs. 2485 ± 88 au; p = 0.0038) (Figure 4C).
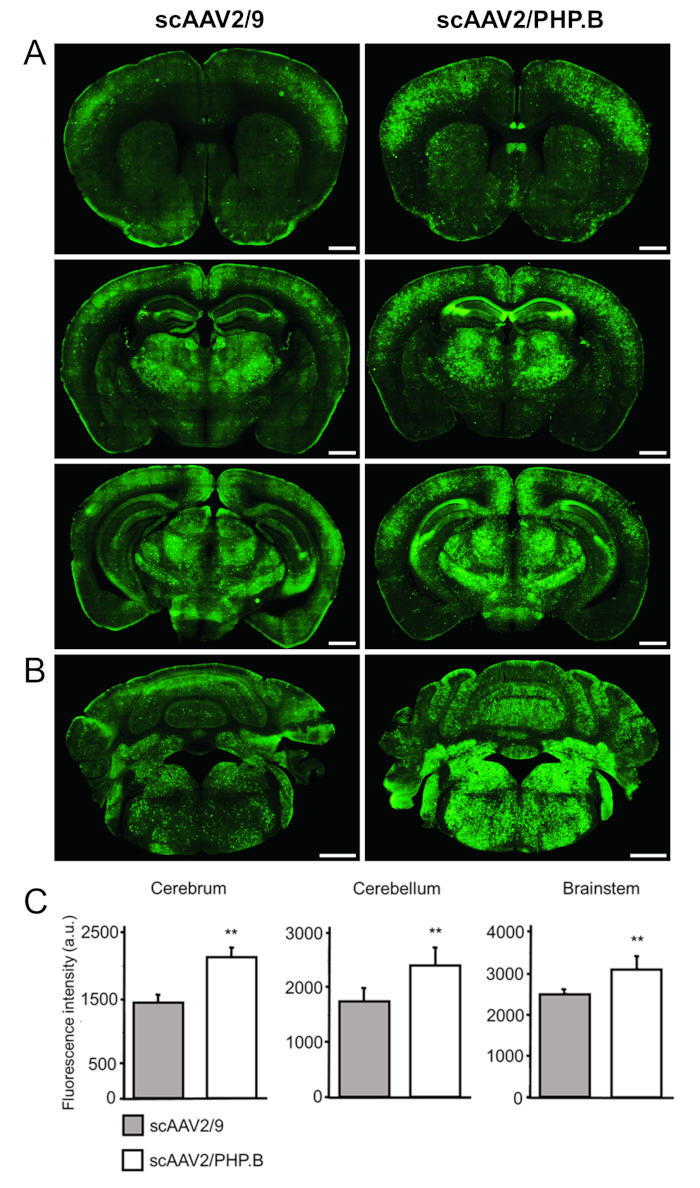
Figure 4: Systemic delivery of scAAV2/PHP.B-CBA-GFP leads to a high GFP expression in the CNS. scAAV2/PHP.B-CBA-GFP or scAAV2/9-CBA-GFP (1 x 1012 vg/mouse) was administered to 6-weeks-old C57/Bl6 mice via tail vein injection. GFP was detected using immunohistochemistry on coronal brain sections 3 weeks postinjection. (A) The cerebrum and (B) the cerebellum and brainstem are shown. Scale bars = 1 mm. (C) The quantification of relative fluorescence intensities was performed to determine the levels of GFP signal achieved with each vector (10 sections per mouse; three mice per vector group). A one-way ANOVA test was performed, followed by a two-tailed Student's t-test; the data are expressed as mean ± standard deviation; **p < 0.01; au. arbitrary units. pCapsid, used for AAV vector production, contains the gene rep from serotype 2 and the gene cap from serotype PHP.B or AAV9, accordingly. This figure has been modified from Rincon et al.32. Please click here to view a larger version of this figure.
