CRISPR-Cas9 technology is a powerful research tool for functional genomic studies. It is rapidly replacing conventional gene editing techniques and has high utility for both genome-wide and individual gene-focused applications. Here, the first individually cloned loci-specific CRISPR-Cas9-arrayed sgRNA library contains 1,070 sgRNAs consisting of sgRNAs targeting 303 random targeting genes, 60 positive controls, 500 non-Human-targeting controls, and 207 CTCF elements or lncRNA targeting genes in four HOX loci (Figure 1, Table 1). This library targets all CTCF core binding motifs, HOX gene associated lncRNAs, known regulatory elements, and several HOX genes as positive controls in the HOX loci. It also contains sgRNAs targeting random non-HOX genes, non-human genes and intergenic regions as negative controls. To enhance efficiency and specificity of CTCF site knock-out (KO) by lentiCRISPR transduction, each targeting site contains 5-10 sgRNAs (Table 1). In the protocol described here, sgRNA libraries are designed according to CTCF binding sites at the HOXA/B/C/D loci and lncRNAs in these loci, which is based on the Broad Institute sgRNA tools (Figure 1, Figure 2). After transduction at a low multiplicity of infection with a MOI of 0.3 in MOLM13 cells carrying the MLL-AF9 fusion, the infection rate is less than one sgRNA/cell followed by puromycin selection, and then the resistant clones grown from seeded single cell were screened for impairment of HOXA9 gene expression.
The workflow for sgRNA library screening was briefly described (Figure 3). First, the virus containing sgRNA library were generated in HEK293T cells with the help of two vectors (psPAX2 and pMD2.G). sgRNA pooled library lentiviruses were concentrated and transduced into MOLM13 AML cells with polybrane (8.0 µg/mL). After a 48 h transduction, cells were treated with the optimal concentration of puromycin. After 5 days, the cells were seeded one cell/well into 96-well plates and the single clones were generated in the presence of puromycin. Finally, sgRNA single clones integrated into genome were identified by one-step RT-PCR, Sanger sequencing and Indel mutation detection (Figure 3). The puromycin resistant single clones are identified through one-step droplet digital RT-qPCR (RT-ddqPCR) according to altering expression of HOXA9 oncogene (Figure 4). Genotyping and Sanger sequence were performed for sgRNA library construction and verification (Figure 2, Figure 4).
sgRNA targeting MOLM13 positive clones in a 96-well PCR plate were further confirmed with the RT-qPCR method based on theexpression levels of HOXA9 genes through comparison with the control cells. Out of the 528 surviving clones screened, 10 clones exhibited more than 50% reduction in HOXA9 levels (Figure 4A). sgRNAs integrated into the HOXA9-reduced, HOXA9-unchanged, and HOXA9-increased clones were further confirmed by PCR amplification of the sgRNA sequences using flanking vector primers. The purified PCR products were ligated into the T vector system through T4 ligase and sent out for identification by Sanger sequence (see step 8). The sequence data indicated that out of 30 clones sequenced, 21 clones included single sgRNA (Table 2). The categories of sgRNA were identified and analyzed according to the HOXA9 expression levels. Six of ten clones showing a reduction in HOXA9 levels contained sgRNAs targeting the CBS7/9 site, but not in the non-human genes, random human genes, and other CTCF site controls (Figure 4 and Table 2).
sgRNA integrated positive single clone-induced Indel mutations are determined by PCR-based genotyping and nuclease digestion based on the nuclease assay (Figure 5). The nuclease digestion assay has been performed to identify Indel mutations occurred in the CBS7/9 boundary in the representative HOXA9-reduced, HOXA9-unchanged, and HOXA9-increased clones. The results revealed that the CBS7/9 mutation has been found in 4 out of the 6 HOXA9-reduced clones: clones #5, 6, 28, and 121, but not in clones #15 and #31 (Figure 5). However, clone #15 contained the sgRNA targeting HOTTIP lncRNA site, while clone #31 contained several sgRNAs targeting HOAIRM1 lncRNA, HOTAIR lncRNA, and HOXD9/10 CTCF binding site (Figures 4B, 5 and Table 2).
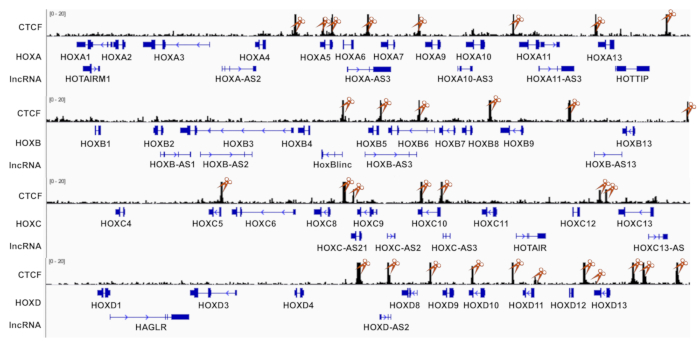
Figure 1: Schematic diagram showing CTCF binding sites and lncRNAs in four HOX gene loci. Each targeting DNA element contains 5-10 different sgRNAs. CTCF ChIP-seq dataset was downloaded from GEO (GSM1335528) and visualized with Integrated Genomic Viewer (IGV). SgRNA targeting CTCF sites in HOX loci were labelled with orange scissors. Please click here to view a larger version of this figure.
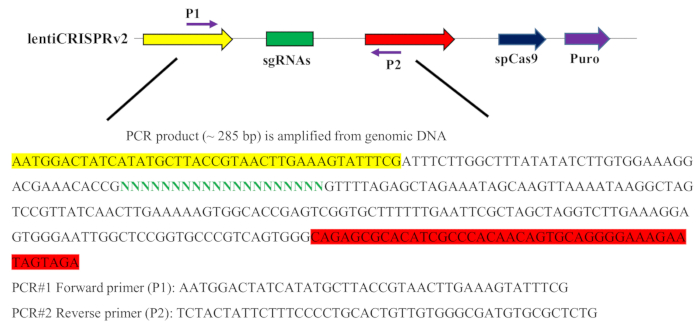
Figure 2: Schematic diagram representing the part of integrating sgRNA vector sequence and PCR amplification primers. The PCR amplification primers were designed according to the blank sequence of the sgRNA lentiviral vector. The forward primer (P1) was highlighted in yellow, the reverse primer (P2) was highlighted in red, and the sgRNA was highlighted in green in the sgRNA lentiviral vector. Please click here to view a larger version of this figure.
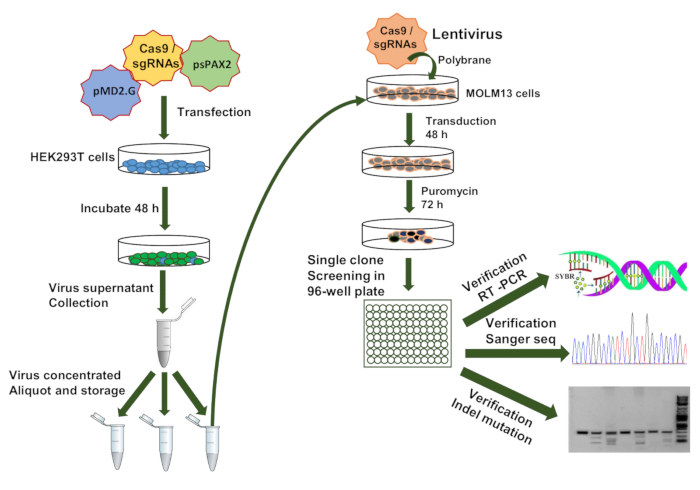
Figure 3: Schematic diagram representing the workflow for sgRNAs library design, construction and verification. This workflow is as follows. First, the sgRNA library was designed and cloned into a lentiviral CRISPR vector, and then the lentivirus was packaged with the sgRNA library lentiviral vector, psPAX2 and pMD2.G vectors in the HEK293T cells. Next, MOLM13 cells were infected with a low MOI (0.3) virus and these cells underwent puromycin selection. Then, the single clone was seeded in a 96-well plate. Finally, the sgRNA single clones integrated into a genome were identified by one-step RT-qPCR, Sanger sequence and Indel mutation detection. Please click here to view a larger version of this figure.
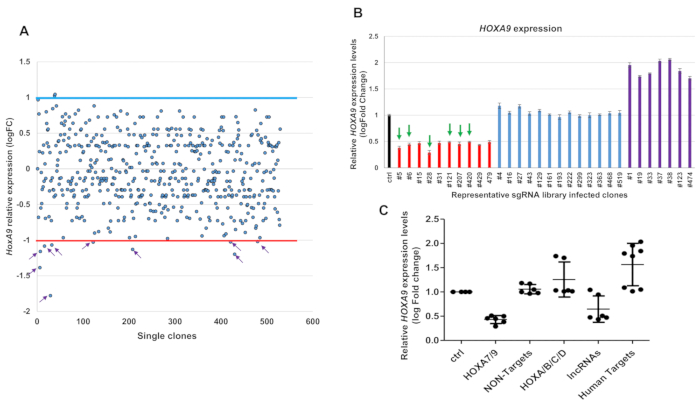
Figure 4: Pooled CRISPR-Cas9 KO library screening identified with one-step RT-qPCR and Sanger sequence. (A) One step RT-droplet digital PCR screening of the HOXA9 expression in single clones infected with lentivirus containing the sgRNA library. The screening of 528 sgRNA library infected clones for HOXA9 expression levels is shown (528 dots). Ten of 528 clones exhibited more than 50% reduction in HOXA9 levels (purple arrows). The red line signifies the boundary of a 2-fold decrease change by comparing with the control cells; the blue line signifies the boundary of a 2-fold increase change. (B) The six clones #5, 6, 28, 121, 207 and 420 were targeted by the CBS7/9 specific sgRNA through Sanger sequence (green arrows). (C) The RT-ddqPCR analysis of HOXA9 levels in WT MOLM13 and the 21 clones containing single targeted sgRNA. The HOXA9 expression data were grouped into five groups in accordance with the categories of sgRNA sequences: HOXA7/9 CTCF site, non-human targets, other CTCF sites in the HOX loci, HOX associated lncRNAs, and other human targets. This figure has been modified from Luo et al.12. For statistics, this data was represented as the mean ± SD from three independent experiments with the Student's t-test. Please click here to view a larger version of this figure.
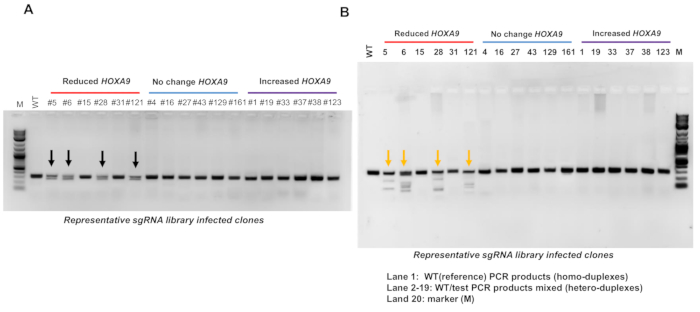
Figure 5: Indel mutations of integrated sgRNAs positive clone confirmed with the PCR-based genotyping and nuclease assay. (A) Genomic DNA was isolated from the representative CRISPR-Cas9 KO library screened clones that exhibited reduced, unchanged, or increased HOXA9 expression levels. The heterozygous deletion of the CTCF site located between HOXA7 and HOXA9 genes (CBS7/9 boundary) was identified by PCR-based genotyping. The HOXA9-decreased clones #5, 6, 28, and 121 exhibited deletion in the CBS7/9 boundary location (black arrows). (B) The Indel mutations in the CBS7/9 site were analyzed by the nuclease digestion assay from the representative clones that exhibited reduced (red line), unchanged (blue line), or increased (purple line) HOXA9 expression levels. The HOXA9-decreased clones #5, 6, 28, and 121 exhibited mutations in the CBS7/9 boundary location (orange arrows). This figure has been modified from Luo et al.12. Please click here to view a larger version of this figure.
Table 1: sgRNAs pool library targeting information. This data is from Luo et al.12. Please click here to download this table.
Table 2: Sangersequencing results of sgRNAs presented in the selected HOXA9-decreased, HOXA9-unchanged, and HOXA9-increased clones. HOXA9-decreased, unchanged and increased clones are highlighted in red, blue and purple, separately. This data is from Luo et al.12. Please click here to download this table.
