IFNγ produced within the first 24 h after Listeria monocytogenes infection is critical to control the spread of this pathogen. Using this protocol, we can visualize not only which cells are producing IFNγ but also whether they are located in a specific microenvironment. To help us delineate the architecture of the spleen, we labeled cells known to have particular location within the spleen. The marker F4/80 labels all macrophages and highlights the red pulp. The marker B220 labels B cells and highlights B cell follicles surrounding the T cell zone. The marker CD169 labels marginal zone macrophages, surrounding the white pulp (Figure 1). Most OTI cells, whether they express IFNγ or not, are present in the white pulp and as such, all images are those of the white pulp, unless indicated.
One critical step in this protocol is the use of BFA to inhibit cytokine secretion. Indeed, the detection of IFNγ by NK cells was greatly impaired when mice were not treated with BFA (Figure 2). Using our protocol, we could find that at least two cell types produce IFNγ 24 h after infection—NK cells and antigen-specific CD8+ T cells (Figure 3)—similarly to what has been found previously by flow cytometry3.
In situ imaging of IFNγ producing cells revealed that IFNγ production is not spread throughout the spleen, but concentrated into discreet areas (Figure 4). Indeed, we found that T cells were activated throughout the spleen (highlighted by T cell clustering), and this did not necessarily correlate with IFNγ production. One likely explanation is that IFNγ production is restricted to the location of the infected cells15,16, and T cell activation—represented by clustering—can be supported by both infected (IFNγ positive) and non-infected (IFNγ negative) antigen-presenting cells. Other stains will be required to pinpoint the exact location and get an indication of the mechanism restricting IFNγ production to this area and its relationship to antigen transfer. Interestingly, we found that activated, clustered, antigen-specific T cells are located throughout the white pulp of the spleen but they produce IFNγ only in regions where NK cells are coexisting with them (Figure 5). As such, the presence of NK cells delineates a specific microenvironment in the white pulp, in which clustered T cells produce IFNγ as opposed to clustered T cells in the other part of the white pulp. This suggests that T cell activation is not sufficient to dictate IFNγ production at this time point.
Another interesting feature highlighted by our protocol is the different sub-cellular localization of IFNγ in NK versus CD8+ T cells5. As shown in Figure 6, while IFNγ localization in NK cells is diffused in the cytosol, CD8+ T cells often recruit IFNγ towards another T cell.
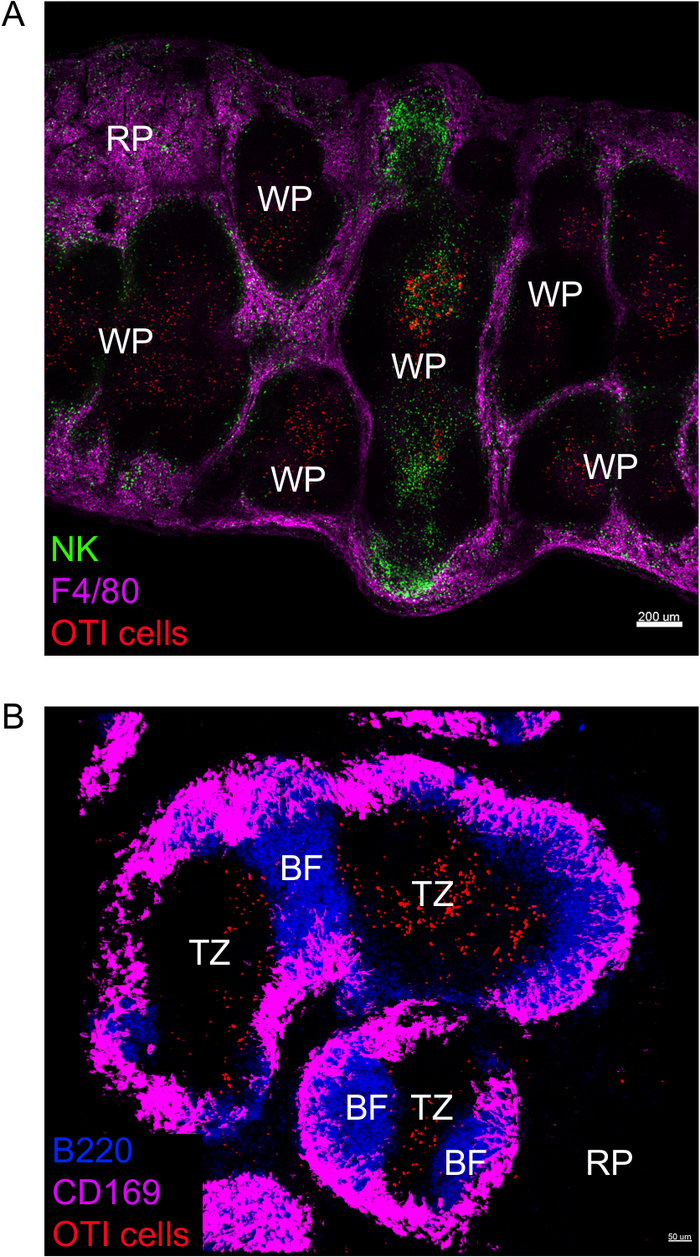
Figure 1: Markers highlighting the spleen architecture. Mice were infected with 2 x 104 CFU LM-OVA and euthanized 24 h post infection. Spleen was explanted and processed as described in the protocol. (A) Sections were stained for NK cells (anti-NCR1 followed by anti-goat IgG-FITC; green), OTI-RFP cells (red) and macrophages (anti-F4/80-APC; magenta). RP = Red Pulp; WP = White Pulp. Scale bar = 200 µm. (B) Sections were stained for B cells (anti-B220-Pacific Blue; Blue), OTI-GFP cells (GFP signal shown in red) and marginal zone macrophages (anti-CD169-Alexa647; magenta). RP = Red Pulp; BF = B cell follicle; TZ = T cell zone. Scale bar = 50 µm. This is a representative image of 3 independent experiments (N = 4). Please click here to view a larger version of this figure.
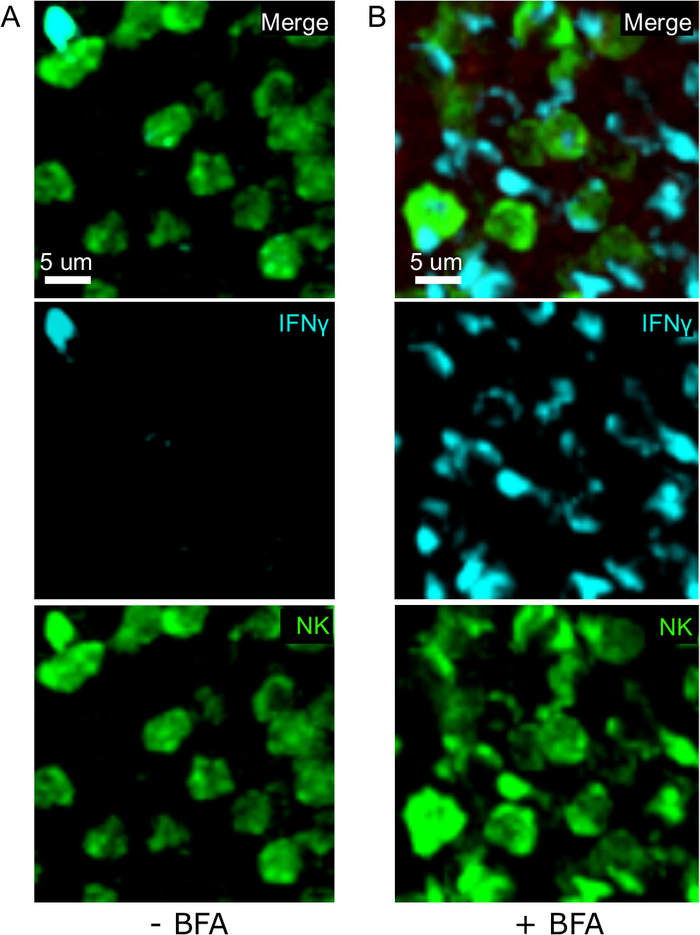
Figure 2: BFA treatment allows for the detection of intracellular IFNγ in situ. Nγ production is restricted to specific areas in the sple Mice were infected with 2 x 104 CFU LM-OVA and treated with BFA (A) or left untreated (B) after 18 h. Mice were euthanized 24 h post infection. Spleen was explanted and processed as described in the protocol. Sections were stained for NK cells (anti-NCR1 followed by anti-goat IgG-FITC; green), OTI-RFP cells (red) and IFNγ (anti-IFNγ-BV421; cyan). Scale bar = 5 µm. This is a representative image of NK cell-rich areas from 3 independent experiments (N = 3). Please click here to view a larger version of this figure.
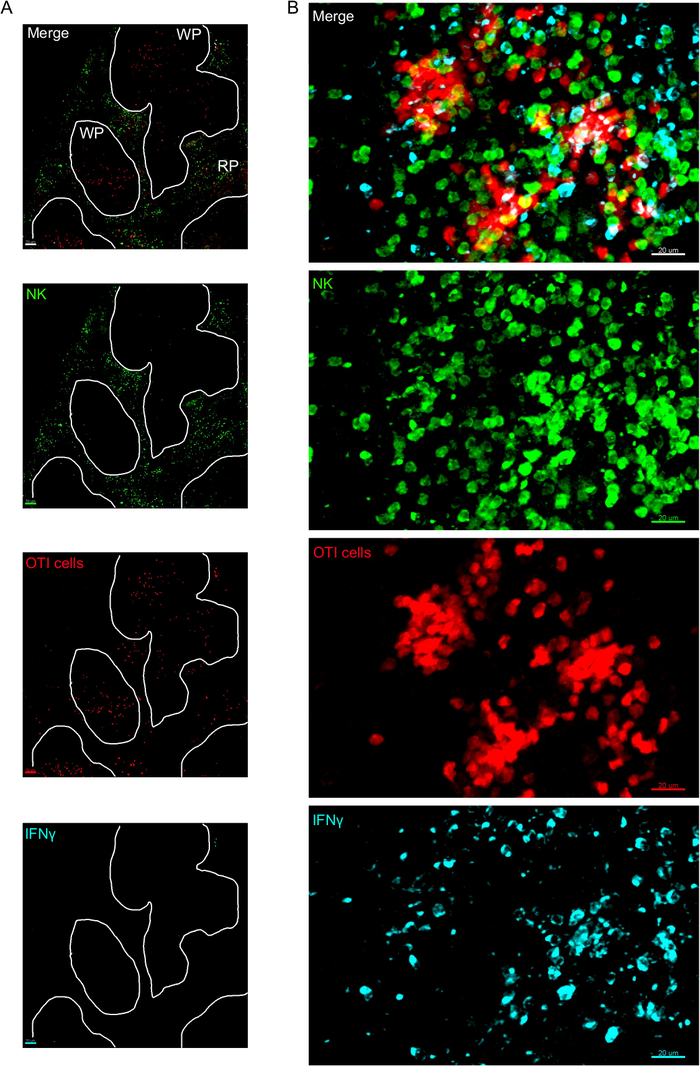
Figure 3: IFNγ producing cells in the spleen. Mice were infected with 2 x 104 CFU LM-OVA when indicated and treated with BFA after 18 h. Mice were euthanized 24 h post infection. Spleen was explanted and processed as described in the protocol. Sections were stained for NK cells (anti-NCR1 followed by anti-goat IgG-FITC; green), OTI-RFP cells (red) and IFNγ (anti-IFNγ-BV421; cyan). (A) Representative image of a spleen from an un-infected naïve mouse to demonstrate absence of IFNγ non-specific staining. White lines delineate the white pulp. WP = White pulp; RP = Red pulp. (B) Representative image of the white pulp from the spleen of a mouse infected by LM-OVA, showing invasion of NK cells to the white pulp and production of IFNγ by NK cells, OTI cells and non-labelled cells. Images are representative from 4 independent experiments (N = 4). Scale bars = 70 µm (A); and 20 µm (B). Please click here to view a larger version of this figure.
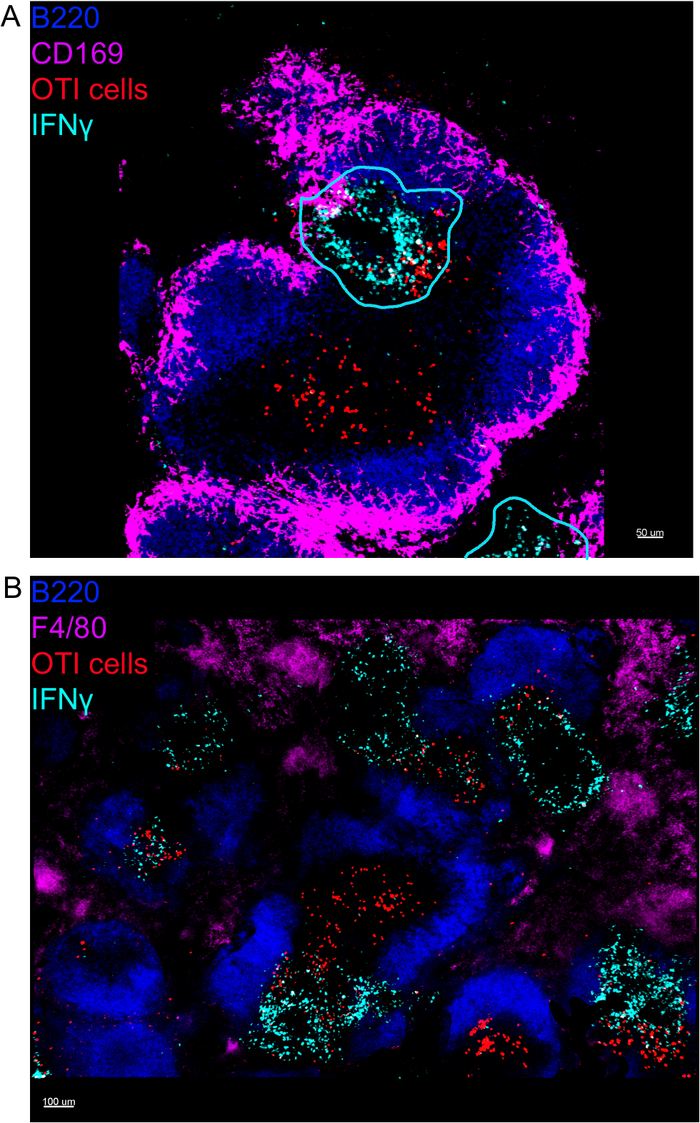
Figure 4: IFNγ production is restricted to specific areas in the spleen following LM-OVA infection. Mice were infected with 2 x 104 CFU LM-OVA and treated with BFA after 18 h. Mice were euthanized 24 h post infection. Spleen was explanted and processed as described in the protocol. All sections were stained for B cells (B220-Pacific Blue Ab, Blue) and IFNγ (anti-IFNγ-biotin followed by streptavidin-PE; cyan). OTI-GFP cells (GFP signal shown in red). Cyan lines correspond to areas of high IFNγ production. Those are representative images of 4 independent experiments (N = 4). (A) Sections were stained for marginal zone macrophages (anti-CD169-Alexa 647, magenta). Scale bar = 50 µm. (B) Sections were stained for all macrophages (F4/80). Scale bar = 100 µm. Please click here to view a larger version of this figure.
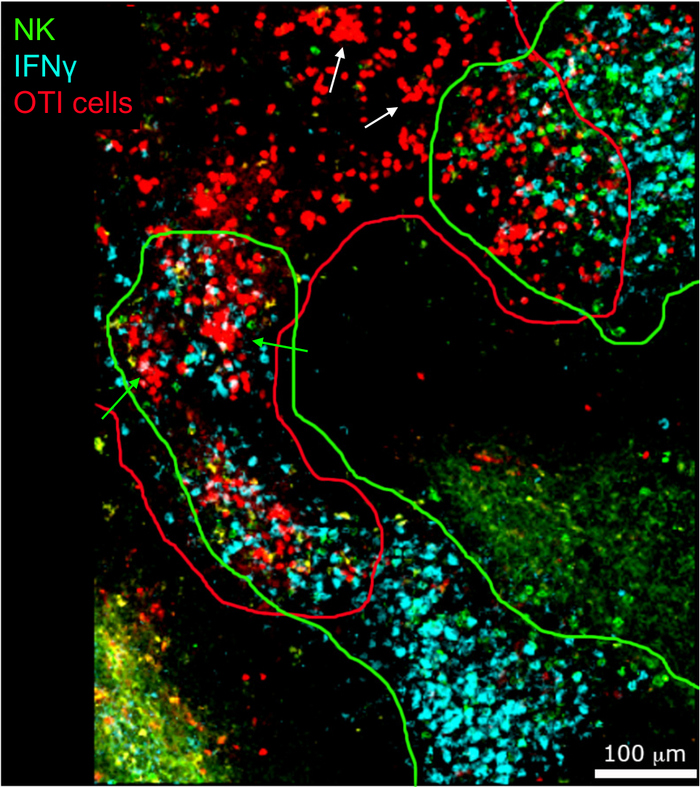
Figure 5: IFNγ production by activated OTI cells occurs in a specific microenvironment. Mice were infected with 2 x 104 CFU LM-OVA and treated with BFA after 18 h. Mice were euthanized 24 h post infection. Spleens were explanted and processed as described in the protocol. Sections were stained for NK cells (anti-NCR1 followed by anti-goat IgG-FITC; green), OTI-RFP cells (red) and IFNγ (anti-IFNγ-BV421; cyan). Green and red lines highlight NK and OTI cell zones, respectively. White arrow indicate examples of T cell clusters not producing IFNγ. Green arrows examples of T cell clusters producing IFNγ. Scale bar = 100 µm. This is a representative image of four independent experiments (N = 4). Please click here to view a larger version of this figure.
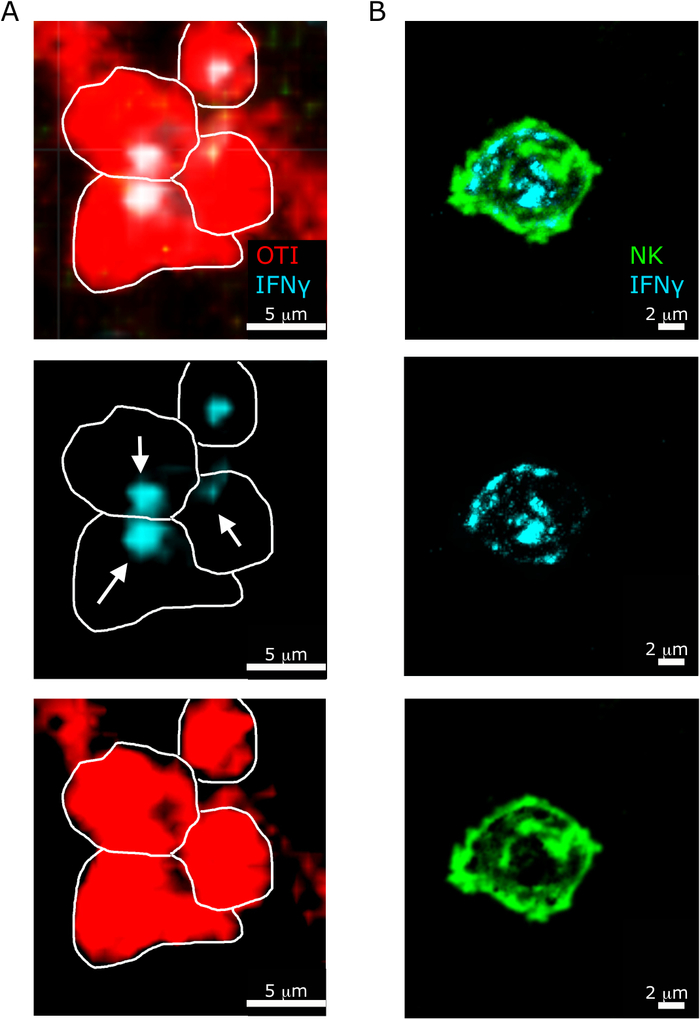
Figure 6: Sub-cellular localization of IFNγ in NK cells and T cells. Mice were infected with 2 x 104 CFU LM-OVA and treated with BFA after 18 h. Mice were euthanized 24 h post infection. Spleen was explanted and processed as described in the protocol. All sections were stained for IFNγ (anti-IFNγ-BV421; cyan). White lines delineate cell edges and white arrows shows directionality of secretion. This is a representative image of two independent experiments (N = 5). (A)- OTI-RFP cells are shown in red. Scale bar = 5 µm. (B) Sections were stained for NK cells (anti-NCR1 followed by anti-goat IgG-FITC; green. Scale bar = 2 µm. Please click here to view a larger version of this figure.
