Generation of In-Frame Gene Deletion Mutants in Pseudomonas aeruginosa and Testing for Virulence Attenuation in a Simple Mouse Model of Infection
Summary
Here, we describe a simple and reproducible protocol of mouse model of infection to evaluate the attenuation of the genetically modified strains of Pseudomonas aeruginosa in comparison to the United States Food and Drug Administration (FDA)-approved Escherichia coli for commercial applications.
Abstract
Microorganisms are genetically versatile and diverse and have become a major source of many commercial products and biopharmaceuticals. Though some of these products are naturally produced by the organisms, other products require genetic engineering of the organism to increase the yields of production. Avirulent strains of Escherichia coli have traditionally been the preferred bacterial species for producing biopharmaceuticals; however, some products are difficult for E. coli to produce. Thus, avirulent strains of other bacterial species could provide useful alternatives for production of some commercial products. Pseudomonas aeruginosa is a common and well-studied Gram-negative bacterium that could provide a suitable alternative to E. coli. However, P. aeruginosa is an opportunistic human pathogen. Here, we detail a procedure that can be used to generate nonpathogenic strains of P. aeruginosa through sequential genomic deletions using the pEX100T-NotI plasmid. The main advantage of this method is to produce a marker-free strain. This method may be used to generate highly attenuated P. aeruginosa strains for the production of commercial products, or to design strains for other specific uses. We also describe a simple and reproducible mouse model of bacterial systemic infection via intraperitoneal injection of validated test strains to test the attenuation of the genetically engineered strain in comparison to the FDA-approved BL21 strain of E. coli.
Introduction
Pseudomonas aeruginosa is an opportunistic bacterial pathogen that can cause life-threatening diseases in humans, especially in the immunocompromised. The pathogenicity of P. aeruginosa is due to the expression of many virulence factors, including proteases and lipopolysaccharide, as well as its ability to form a protective biofilm1. Because of its ability to produce virulence factors and cause disease in humans, using P. aeruginosa to make commercial products presents safety concerns. Nonpathogenic strains of E. coli have traditionally been used to bioengineer medical and commercial products for human use. However, some products are difficult for E. coli to make, and many are packaged in inclusion bodies, making extraction laborious. Engineered bacterial strains with the ability to make and secrete specific products is highly desirable, as secretion would likely increase yield and ease purification processes. Thus, nonpathogenic strains of other species of bacteria (e.g., species that utilize more secretion pathways) may provide useful alternatives to E. coli. We recently reported the development of a strain of P. aeruginosa, PGN5, in which the pathogenicity and toxicity of the organism is highly attenuated2. Importantly, this strain still produces large quantities of the polysaccharide alginate, a commercially interesting component of the P. aeruginosa biofilm.
The PGN5 strain was generated using a two-step allelic exchange procedure with the pEX100T-NotI plasmid to sequentially delete five genes (toxA, plcH, phzM, wapR, aroA) known to contribute to the pathogenicity of the organism. pEX100T-NotI was generated by changing the SmaI to a NotI restriction enzyme recognition site within the multiple cloning site of the plasmid pEX100T, which was developed in Herbert Schweizer's lab3,4. The recognition site for the restriction enzyme NotI is a rarer DNA sequence compared to SmaI and less likely to be present in sequences being cloned, thus it is more convenient for cloning purposes. The plasmid carries genes that allow for selection, including the bla gene, which encodes ß-lactamase and confers resistance to carbenicillin, and the B. subtilissacB gene, which confers sensitivity to sucrose (Figure 1A). The plasmid also carries an origin of replication (ori) compatible with E. coli, and an origin of transfer (oriT) that allows for plasmid transfer from E. coli 到 Pseudomonas species via conjugation. However, the plasmid lacks an origin of replication compatible with Pseudomonas, and thus cannot replicate within Pseudomonas species (i.e., it is a Pseudomonas suicide vector). These characteristics make pEX100T-NotI ideal for targeting genetic deletions from the Pseudomonas chromosome. Plasmid cloning steps are carried out using E. coli and the resultant plasmid is transferred to Pseudomonas by transformation or conjugation. Then, through homologous recombination events and selective steps, the targeted in-frame deletion is generated, marker-free. This method of sequentially deleting genomic regions from the chromosome of P. aeruginosa could be used to generate highly attenuated Pseudomonas strains, like PGN5, or to design strains for other specific uses (e.g., strains deficient in endonucleases for plasmid propagation or strains deficient in proteases for production of proteins of interest).
The overall virulence of strains of bacteria is affected by growth conditions and phases, during which mutations occur frequently. Therefore, measuring the safety of genetically-engineered strains can be challenging. To evaluate bacterial isolates for systemic virulence, we adapted a previously published protocol of infection by intraperitoneal injection of C57BL/6 mice5. We modified this procedure to use frozen bacterial stocks for injection, which allowed for precise dosing and easy validation of the strains used. In this model, the E. coli strain BL21, which has been FDA-approved for production of biopharmaceuticals, was used as a control safety standard for determining the relative pathogenesis of the strain6,7,8. The main advantage to using this method is that it is reproducible and minimizes sources of variation, as infecting strains are validated for bacterial cell number, phenotype, and genetic markers both before and after infection. With these controlled steps, the number of animals required is reduced. In this model, P. aeruginosa strains that result in C57BL/6 murine mortality rates equal to or less than E. coli BL21 when injected intraperitoneally may be considered attenuated. This simple mouse model of infection may also be used to assess the attenuated pathogenicity of genetically engineered strains from other species using the FDA-approved E. coli strain as the reference. Steps 1-7 detail the generation of sequential genomic deletions in P. aeruginosa (Figure 1) and steps 8-12 detail the use of a mouse model to test the pathogenicity of P. aeruginosa strains.
Protocol
Before beginning animal experiments, the protocol to be used must be approved by the Institutional Animal Care and Use Committee (IACUC). Approval for the protocol described was obtained through the IACUC at Marshall University (Huntington, WV, USA).
1. Plasmid Design
- To generate a genetic deletion using the pEX100T-NotI plasmid, clone the regions of DNA flanking the desired deletion sequence and insert into the NotI restriction site of the plasmid. The plasmid insert should contain about 500 nucleotides upstream of the target sequence directly adjacent to about 500 nucleotides downstream of the target deletion sequence. Additionally, the insert should contain the NotI recognition sequence (GCGGCCGC) at its 5' and 3' ends (Figure 1B).
2. Plasmid Preparation
- Option 1: Utilize traditional cloning procedures. Use PCR to amplify genomic regions upstream and downstream of the gene of interest, followed by crossover PCR9,10 to join the generated fragments, restriction endonuclease digestion of the PCR product and plasmid, and ligation11 (Figure 1B,C).
- Option 2: After designing the deletion sequence in silico, contract a company that de novo synthesizes it to insert into the plasmid pEX100T-NotI. Many companies have streamlined the process of cloning to quickly and efficiently generate the plasmid of interest. Additionally, sequence verify the plasmids to be mutation-free prior to delivery.
3. E. coli Transformation
- Transform electrocompetent E. coli with the plasmid according to the manufacturer's recommendations. Using a sterile inoculating loop, streak 10 µL of the transformation reaction for isolated colonies onto a pre-warmed Luria Broth (LB) agar plate supplemented with 100 µg/mL of carbenicillin and incubate overnight at 37 °C.
NOTE: All equipment and media used to culture bacteria should be treated according to the institution's safety guidelines. - Passage twice.
- Remove the plate from the incubator and identify an isolated colony. Using a sterile inoculating loop, pick up the colony and streak a pre-warmed LB agar plate supplemented with 100 µg/mL of carbenicillin for isolated colonies. Incubate overnight at 37 °C. Repeat this step once more to generate a pure culture.
- Using a sterile inoculating loop, inoculate 5 mL of LB with a single colony from the final agar plate. Place the culture in a shaking incubator at 37 °C overnight. The next day, mix 1 mL of this culture with 1 mL of 5% in a cryovial and store at -80 °C to generate a frozen stock of the strain.
4. Bacterial Strain Preparation and Triparental Conjugation
- Use a single isolated colony from agar plates of the following strains to inoculate broth cultures and place in a shaking incubator overnight at 37 °C.
- Add E. coli pEX100T-NotI into 5 mL of LB supplemented with 100 µg/mL of carbenicillin.
- Add P. aeruginosa strain PAO1 into 5 mL of Pseudomonas Isolation Broth (PIB).
- Add E. coli prk2013 into 5 mL of LB supplemented with 50 µg/mL of kanamycin.
NOTE: The prk2013 plasmid is a helper plasmid that replicates in E. coli but not P. aeruginosa; it carries the trans-acting transfer genes that mobilize the pEX100T-NotI plasmid from the E. coli donor to the P. aeruginosa recipient12. P. aeruginosa is a Biosafety Level 2 (BSL-2) pathogen. Please follow the institution's guidelines for safety when working with BSL-2 organisms.
- The next day, remove overnight cultures from the incubator and add 0.5 mL of each culture to a 1.5 mL microcentrifuge tube. Centrifuge at 6,000 x g for 5 min. Discard the supernatant and suspend the cell pellet in 50-100 µL of LB.
- Pipette the entire cell suspension in one droplet onto a pre-warmed LB agar plate. Allow the droplet to dry. Then invert the plate and incubate at 37 °C for 4-6 h.
- After the incubation, use a sterile inoculation loop to collect the cells into 1 mL of LB in a microcentrifuge tube. Pipette up and down to mix the cells.
- Using a cell spreader, streak cells evenly onto a dry pre-warmed Pseudomonas Isolation Agar (PIA) plate supplemented with 300 µg/mL of carbenicillin. Streak multiple plates with increasing volumes of the cell mixture (e.g., 10 µL, 100 µL, 500 µL). Incubate overnight at 37 °C.
5. Detection of Single-crossover Recombinants of P. aeruginosa
- Remove plates from the incubator and inspect for isolated carbenicillin-resistant colonies. Because the pEX100T-NotI plasmid cannot replicate in P. aeruginosa, colonies that grew on carbenicillin-supplemented plates should have arisen from cells in which the plasmid was integrated into the chromosome.
- Choose at least 4 of these colonies and streak for isolation onto pre-warmed plates of PIA supplemented with 300 µg/mL of carbenicillin. Incubate plates overnight at 37 °C.
- Remove plates from the incubator and inspect for growth. Carbenicillin-resistant colonies should be single-crossover recombinants (i.e., they have incorporated the plasmid into the chromosome via a recombination event between a homologous region of the plasmid insert and the chromosome of P. aeruginosa).
- Patch 8 or more colonies with sterile toothpicks onto pre-warmed plates of: 1) PIA supplemented with 300 µg/mL of carbenicillin and 2) PIA supplemented with 300 µg/mL of carbenicillin and 10% sucrose (without glycerol).
- If no colony growth was obtained from step 5.1, repeat the conjugation and increase the volume of the cell mixture streaked in step 4.5. If too much growth occurred, repeat conjugation and decrease the volume streaked.
- If the conjugation repeatedly fails, prepare electrocompetent cells of the P. aeruginosa strain and transform directly with the pEX100T-NotI plasmid. Detailed protocols for preparation of electrocompetent P. aeruginosa and transformation are available elsewhere13,14.
- Incubate plates at 37 °C overnight.
- Remove plates from incubator and inspect for growth. True single-crossover recombinants will be carbenicillin-resistant and sucrose-sensitive (i.e., colonies that grew on PIA supplemented with carbenicillin, but did not grow on PIA supplemented with carbenicillin and sucrose).
- Choose 4 or more true single-crossover recombinants and inoculate each into 5 mL of LB without selection. Incubate in a shaking incubator at 37 °C overnight.
- If no single-crossover recombinants were detected, repeat the conjugation.
6. Detection of Double-crossover Recombinants of P. aeruginosa
- For each broth culture, inoculate 10 µL of culture onto a pre-warmed plate of PIA supplemented with 10% sucrose (without glycerol) and streak for isolated colonies. Incubate plates overnight at 37 °C.
- The next day, remove plates from the incubator and inspect them for growth. Sucrose-resistant colonies should be double-crossover recombinants (i.e., have removed the plasmid from the chromosome via a recombination event between the other homologous region of the plasmid insert and the P. aeruginosa chromosome).
- Patch at least 20 colonies with sterile toothpicks onto pre-warmed plates of: 1) PIA, 2) PIA supplemented with 10% sucrose (without glycerol), and 3) PIA supplemented with 300 µg/mL of carbenicillin.
- Incubate plates overnight at 37 °C.
- Remove plates from the incubator and examine them for growth. True double-crossover recombinants will be carbenicillin-sensitive and sucrose-resistant (i.e., colonies that grew on PIA and PIA supplemented with sucrose, but did not grow on PIA supplemented with carbenicillin).
7. Gene Deletion Confirmation via Colony PCR
- Prepare 10-20 colonies for a deletion screen with PCR.
- Pick up the growth from a suspected double-crossover recombinant with a sterile toothpick and suspend cells in 50 µL of 1x phosphate buffered saline (PBS). Boil suspension at 100 °C for 10 min, centrifuge for 3 min at 13,000 x g, and then place on ice.
- Perform PCR to screen colonies for the targeted deletion.
- Use 1 µL of the supernatant as the template in a 25 µL PCR reaction to confirm deletion of the gene of interest.
- Use gene-specific primers that amplify the region of the genomic deletion. Use primers that amplify the region of the genomic deletion plus 100-200 bp of flanking upstream and downstream sequences.
- Prepare a separate control PCR reaction with the parent strain (e.g., PAO1).
NOTE: Thermocycler conditions will vary depending on the optimal annealing temperature for primer pairs, the polymerase cocktail used, and the length of the region to be amplified.
- Perform agarose gel electrophoresis on the PCR products. Colonies in which the region of interest has been deleted yield smaller amplification products than colonies that lack the deletion (Figure 2).
- Choose one or more colonies with the PCR-confirmed deletion. Streak for isolated colonies onto a pre-warmed PIA plate(s) and incubate at 37 °C overnight.
- Passage at least one more time. Remove plate(s) from the incubator and identify an isolated colony. Using a sterile inoculating loop, pick up the colony and streak a pre-warmed PIA agar plate for isolated colonies. Incubate overnight at 37 °C.
- Choose a colony from each final plate and use to inoculate 5 mL of PIB. Place in a shaking incubator at 37 °C overnight.
- Mix 1 mL of this culture with 1 mL of 5% skim milk in a cryovial and store at -80 °C to generate a stock of the strain.
- Using this culture, prepare genomic DNA from the strain (e.g., using a DNA purification kit). Amplify the genomic deletion region using PCR and primers specific to the region of interest.
- Purify these PCR products (e.g., with a DNA purification kit, or phenol-chloroform extraction) and either sequence directly with the gene-specific primers or ligated into a vector for sequencing with plasmid-specific primers.
- After the gene deletion is confirmed through sequencing, repeat this procedure with the new deletion strain to sequentially generate numerous marker-free genomic deletions. When the desired strain is generated, use whole genome sequencing to verify the targeted deletions and to detect other changes to the genome (compared to the reference strain, e.g., PAO1) that occurred throughout the process. After annotating the genes, deposit the sequence to GenBank and record accession numbers.
8. Preparation of Bacterial Strain for Animal Testing
- To test for attenuated pathogenicity of P. aeruginosa strains, first prepare validated cultures and stocks. Prepare the P. aeruginosa strains of interest, a wild-type strain of P. aeruginosa (virulent), and an FDA-approved strain of E. coli (e.g., BL21) to serve as a nonpathogenic safety control.
- Streak the strains of the bacteria being tested onto selective agar from sequenced and validated frozen stocks. Incubate at 37 °C overnight.
- With a sterile inoculating loop, pick up a single colony from each strain and streak for isolated colonies onto selective media again. Incubate at 37 °C overnight.
- Remove plates from the incubator. For each strain, choose a single colony and streak for isolation onto LB plates.
- After 24 h of growth at 37 °C, inoculate a 500 mL flask containing 250 mL of LB with a single colony isolate from each strain.
- Validation step: using the remnants of the same colony, validate the strain using PCR and strain-specific primers, and/or primers to verify the presence of genetic modifications made to the strain. Use the primers below for verification of strains in the example presented:
E. coli BL21:
T7 polymerase F:TGGCTATCGCTAATGGTCTTACG
T7 polymerase R:TTACGCGAACGCGAAGTCC
VE2 and PGN5:
aroA F: GCGAACGCCAACAGCCGATAAAGC
aroA R: ATCTGGCTCGCGATGCCGGTCC
- Validation step: using the remnants of the same colony, validate the strain using PCR and strain-specific primers, and/or primers to verify the presence of genetic modifications made to the strain. Use the primers below for verification of strains in the example presented:
- Incubate the cultures in a shaking incubator at 160 rpm and 37 °C until they reach log phase growth (i.e., OD600 measurement of 0.4-0.6 on a spectrophotometer).
- Using the OD600 value obtained when log phase was achieved, calculate the volume of broth required to yield 2.5 x 109 colony forming units (CFU) per mL. Pellet the volume of broth calculated in 50 mL tubes at 4,500 x g for 10 min.
- Discard the supernatant and resuspend the pellet in one tube using 50 mL of 1x PBS to wash the cells. Centrifuge again at 4,500 x g for 10 min.
- Discard the supernatant and resuspend the pellet in 25 mL of 5% skim milk in 1x PBS.
- Validation step: Use a sample of the 25 mL resuspension to perform viable plate counts to determine the number of CFU/mL.
- Aliquot the 25 mL skim milk culture resuspension into 2 mL culture stocks in 2 mL cryovials. Flash freeze in liquid nitrogen and store at -80 °C at least overnight before use.
9. Validation of Growth and Strain of Stocks Stored for Animal Testing.
- For each strain to be tested, remove at least 3 cryovials of frozen stocks from -80 °C storage and thaw at 4 °C for 2-4 h. If any frozen stock remains, briefly warm at 37 °C.
- Take small samples from each cryovial to validate each strain.
- Perform viable plate counts to determine the number of CFU/mL. It is normal to have fewer CFU/mL after freezing, due to death of some bacterial cells.
- Use PCR and strain-specific primers to validate each strain.
- Streak each strain onto selective media to verify the phenotype.
- After confirming that strains are of the correct genotype and phenotype, and validating CFU/mL, proceed to animal testing.
10. Inoculation of Animals with Bacterial Strains by Injection
- On the morning of injections, remove cryovials of the bacterial strains being tested and thaw at 4 °C for 3-4 h. Thaw 0.5 mL for each mouse that will be injected. Keep vials on ice after thawing and inject mice within 2 h.
- After thawing, transfer each cryovial to a new 2 mL tube and centrifuge at 4,500 x g for 10 min, discard the supernatant, and resuspend the cell pellet in 1 mL of 1x PBS.
- Centrifuge again at 4,500 x g for 10 m. Discard supernatant and resuspend pellet in 1x PBS to a final concentration 2.5 x 109 CFU/mL. To determine the amount of 1x PBS needed for resuspension, use the CFU/mL data obtained from viable plate counts on frozen stocks in step 2.2.1. The exact amount of 1x PBS used will vary slightly between strains.
- Take 3 samples from final suspension of each strain to validate CFU/mL, genotype, and phenotype as described above.
- For each strain, aliquot 1.5 mL of PBS/cell suspension into one 2 mL tube per 5 mice to limit the number of times the tube is entered. Also, prepare tubes of 1x PBS for control injections.
- Gather mice (10 male and 10 female C57BL/6 per group for this experiment) and materials needed for injections (syringes, needles, sharps containers, markers, pen and paper, etc.). Move to the sterile animal surgical room. Wipe all surfaces with sanitizing wipes.
- To eliminate distress and risk of injury to experimenter, only bring one sex and experimental group of mice to the surgical room at a time (e.g., a group of 10 male mice to be infected with a particular strain). Wear two pairs of latex gloves to eliminate puncture of gloves if bitten. Wear lab coat, safety glasses, and face mask to avoid contamination.
- Begin injections of the control group with 1x PBS. This will ascertain whether any adverse effects result from injection alone.
- Remove a mouse from the cage. Only remove one mouse at a time.
- Weigh the mouse and mark its tail with permanent marker to track for weight loss post-injection.
- Open a new 1 mL syringe and 27 G needle (use a new syringe and needle for each mouse to eliminate contamination) and inject 200 µL of sterile 1x PBS.
- Grab the mouse behind its ears using the thumb and forefinger. Pinch to create skin fold at nape of neck to hold onto – a tighter fold reduces neck movement and risk of being bitten during injection. Secure tail into the palm using the pinky to hold mouse flat with little movement.
- Turn the mouse over and insert needle at 30° angle into the peritoneal cavity to the left or right side of midline. Lift the needle slightly once inserted to ensure it was inserted into the intraperitoneal area and not into organs. Slowly inject the PBS and then withdraw the needle.
- Place the used needle in a designated biohazard sharps container. Do not re-use the syringe or needle. A bolus at the site of injection is typical.
- After injection, move mouse to a separate cage.
- Repeat the procedure with the next mouse. After all the mice of one cage are injected, return them to their original cage immediately.
- After injecting the control group, begin injecting suspensions of strains to be tested following same procedure.
- Inject 200 µL of the cell suspension. When beginning with cell suspensions of 2.5 x 109 CFU/mL, each mouse receives 5 x 108 CFU.
NOTE: These concentrations were optimized with preliminary animal research to determine lethal doses of each strain. The dosing may need to be adjusted for other strains/species.
- Inject 200 µL of the cell suspension. When beginning with cell suspensions of 2.5 x 109 CFU/mL, each mouse receives 5 x 108 CFU.
- Once all injections are complete, return mice to the housing room to alleviate distress. Clean work area with sanitizing wipes.
- Monitor the animals for mortality following injection by checking cages for dead mice every 3 h for 72 h and every 12 h for 10 days. Record the weight of mice every 12 h to determine weight loss due to illness.
- Record adverse behavior in the hours following injection, such as difference in posture, lack of grooming or burrowing, immobility, or changes in breathing. Mice that decease from injury associated with injection will exhibit adverse behavior and die quickly following injection. On the other hand, mice that decease from infection will not start to exhibit adverse behavior or death until after 18 h. Any mice that do exhibit adverse behavior or extensive signs of morbidity including rapid or labored respiration, immobility, hunched posture, sunken eyes, severe dehydration, or others should be euthanized to reduce unnecessary suffering (as compliant in our approved IACUC protocols). However, in our experiments, euthanization was not required for any mice during the experiment. Surviving mice were euthanized 10 days following completion of testing.
- Following the 10-day monitoring period, allow the animals remaining to recover fully and become clear of any infection administered during the testing. Euthanize animals following IACUC procedure.
11. Statistical Analysis of Animal Mortality
- Perform statistical analysis using graphing software. Any software capable of producing graphs and performing statistical analysis is suitable.
- To plot the mortality data, use the X column to represent time (h) and Y column to represent the groups tested.
- Represent each mouse or subject in the study using code = 0 (zero) or code = 1, indicating survival or death, respectively.
- For each animal that dies, place a 1 in the Y column of that group at the time of death on the X column. If there are multiple deaths at a single time point within a group, a copy of that time point can be placed in the X column. For example, if three subjects within a group die at 3 h, the 3 h time point will appear three times in the X column.
- For all surviving animals within a group, place a zero in the Y column at the final time point measured. For example, if four mice survive, place four ending time points in the X column marked with four zeros in the Y column.
- After animal data is entered for all groups, use a survival graph template to produce a survival graph.
- Leave default coding as 0 and 1.
- Set the parameters for the graph as percentage.
- Once parameters for survival curve are selected, perform statistical analysis using a Mantel-Cox (log rank) test.
- Format Kaplan-Meier plots using statistical data.
NOTE: Strains that exhibit mortality rates that are less than or equal to the parent strain and the FDA-approved strain (e.g., E. coli BL21) may be considered attenuated.
12. Visualization of the Infection with Bioluminescence
- To visualize the progress of the infection, insert a chromosomal bioluminescent operon (luxCDABE) into the PGN5 and VE2 strain tested. The plasmids and protocol used to label these strains were developed in the Schweizer lab and may not be compatible with all species/strains of bacteria13. Importantly, visualization of the infection is optional; thus, genomic insertion of this operon is not necessary to perform the mortality study described above.
- Prepare and validate strains using the method described above. Additionally, check for bioluminescence in labeled strains at each validation step.
- After strains are prepared, inject the animals in groups of 10 with the bioluminescent strains following the steps above.
- Image the animals every 3 h for 24 h using an animal imaging system capable of bioluminescence.
- First prepare the imager by setting the camera parameters and heating the stage for the animals. Also set the oxygen flow to 1.5 L per min (or following manufacturer's recommendations).
- After the imager and stage are stabilized, place one mouse into the anesthesia chamber immediately following injection and administer 3.5% isoflurane into the chamber with O2 flow for about 4 min. The anesthesia methods may vary depending upon the chamber and/or anesthetic agent used; follow the manufacturer's recommendations. Determine proper anesthesia via withdrawal reflex test.
- Move the mouse to the temperature stabilized stage. Position the mouse on its back with arms outstretched and fit the mouse with a nose cone for administration of 2.5% isoflurane throughout the imaging procedure.
- Close the door and take bioluminescent images and X-rays of mouse.
- When imaging is complete, return the mouse to its cage and monitor it. The mouse should regain consciousness within 3-5 min.
- Continue to image mice every 3 h for 24 h, each time using a different mouse from each group. Do not reimage a mouse within 24 h due to the possibility of adverse effects due to re-exposure to anesthesia. A single mouse should only receive one dose of anesthesia every 24-36 h. Clean the imaging platform after each mouse is imaged. Turn off the imager between imaging time points.
NOTE: Bioluminescence will fade, regardless of the strain injected. The intensity and longevity of bioluminescence will vary depending on many factors, including the number of bacteria injected, mouse strain, bacterial strain, etc.
Representative Results
As shown in Figure 2, the targeted genomic deletion can be confirmed using colony PCR with specific primers that amplify the region of interest. Colonies that carry a genomic deletion will yield a shorter PCR band size in comparison to wild-type colonies. A PCR-screen of 10-12 colonies is usually sufficient to detect at least one colony that carries the targeted deletion. If no deletions are detected after multiple rounds of screens, repeat the procedure beginning with the conjugation. If the deletion still fails, the plasmid insert may need to be confirmed through sequencing, redesigned, or the deletion may be lethal. Upon the verification of a gene deletion via PCR, confirm the deletion through sequencing. The resulting strain may be subjected to the procedure repeatedly to generate sequential genomic modifications.
As shown in Figure 3, mortality associated with intraperitoneal injection of the attenuated strain of P. aeruginosa PGN5 (+mucE) was 0%, which was equivalent to mortality observed with E. coli BL21. On the other hand, intraperitoneal injection of the parent strain (VE2) was fatal to 80% of mice. These results were obtained with extensive steps to validate the strains injected. While the exact cause of death in these mice is unknown, it can at least in part be attributed to the expression of virulence factors in the parent strain that were deleted from the attenuated PGN5 strain. Differences in the infection progression was tracked using bioluminescence-marked parent and attenuated strains. The attenuated strain remained localized at the site of injection until bioluminescence faded (Figure 4). The clearance of the infection most likely coincided with the fading of the bioluminescence. Bioluminescence was not detected 24 h after injection and mice lived for weeks following injection until sacrificed, with no adverse effects observed.
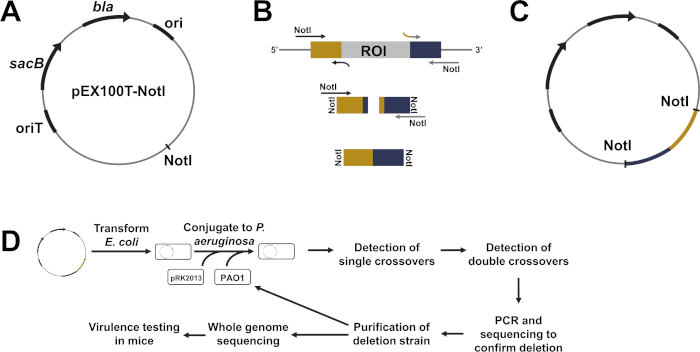
Figure 1: Generating gene deletions in P. aeruginosa with pEX100T-NotI. (A) Map of the pEX100T-NotI plasmid. (B) Generation of a construct composed of regions directly upstream (yellow) and downstream (blue) of the region of interest (ROI), flanked with NotI restriction enzyme recognition sites. First, PCR-amplify upstream and downstream regions independently with specific primers that add 5' NotI digestion sites (e.g., NotI-aroA F CGCGGCCGCTGAAGGTCCTGGGCTCCTATCCGAAAGCGGTGCTCT and NotI-aroA R GCGGCCGCAGTTGGGTTGTTCTGCGATGGCGCCAGGCA) and 3' overlapping homologous regions as shown (e.g., aroA-crossover F CTCCAGGCGCTGGGCAAGGTGCTGGCGCATGACTGAGGTCACGCCGGTCGCCGTGGAGAACA and aroA-crossover R TGTTCTCCACGGCGACCGGCGTGACCTCAGTCATGCGCCAGCACCTTGCCCAGCGCCTGGAG. Then, use PCR withNotI-containing primers to join the upstream and downstream products generated in the first PCR reaction. (C) The pEX100T-NotI plasmid, armed and ready. Ligate the NotI-digested cross-over PCR product into the NotI-digested plasmid. (D) Flow diagram of the process to delete genomic regions from the P. aeruginosa chromosome using the pEX100T-NotI plasmid. After the desired deletion has been confirmed and purified, the resultant strain can be taken through the procedure repeatedly to delete other genomic regions from the chromosome. When the desired strain is obtained, sequence the whole genome to confirm deletions and other changes to the chromosome. The pathogenicity of the strain can then be tested in mice using the procedure outlined in Part II of the Protocol. Please click here to view a larger version of this figure.
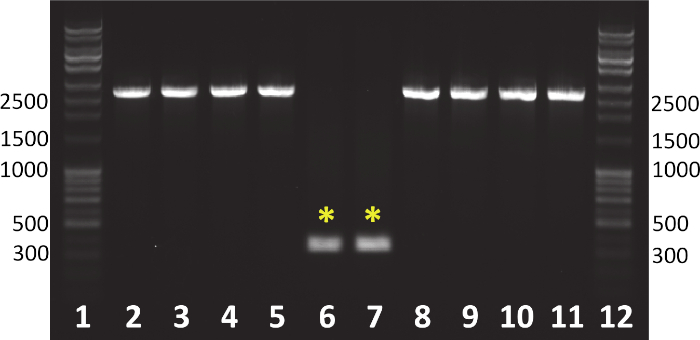
Figure 2: Gel electrophoresis of colony PCR products from a screen for aroA deletion to generate the attenuated P. aeruginosa strain, PGN5. Colony PCR products run in lanes 2-5 and 8-11 indicate colonies with wild-type aroA. Colony PCR products run in lanes 6 and 7 carry the aroA gene deletion, indicated by the smaller PCR product (yellow asterisks). Primers used specifically amplified the genomic region containing the aroA gene: aroA-F: GCGAACGCCAACAGCCGATAAAGC, and aroA-R: ATCTGGCTCGCGATGCCGGTCC. Expected PCR product size in wild-type colonies was 2,548 nucleotides (nt). Expected PCR product size in colonies with aroA deletion was 307 nt. A DNA ladder was run in lanes 1 and 12. Please click here to view a larger version of this figure.
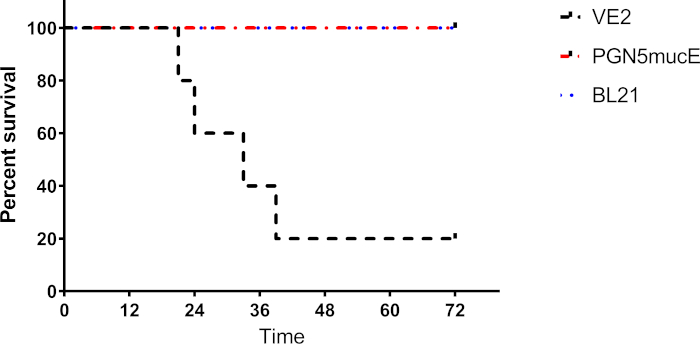
Figure 3: Overall mortality of mice injected with pathogenic P. aeruginosa strain (VE2), attenuated P. aeruginosa strain (PGN5+mucE), and FDA control E. coli strain (BL21). Only mice injected with pathogenic parent strain exhibited mortality at 80%. Attenuated P. aeruginosa strain and FDA control E. coli strain exhibited 0% mortality. Please click here to view a larger version of this figure.
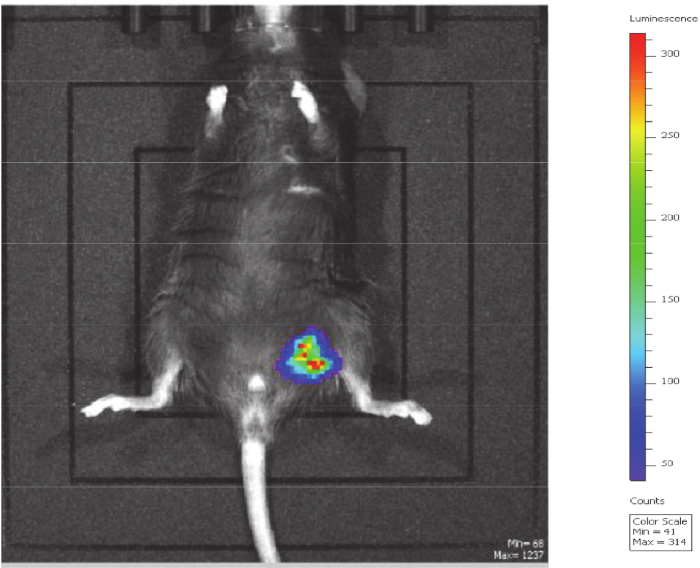
Figure 4: Image of mouse 3 h post-injection of attenuated strain of P. aeruginosa PGN5+mucE carrying a bioluminescent marker. The bioluminescent bacteria were detectable until 18-24 h following injection. During this period, the bioluminescence remained at the site of injection indicating the bacteria stayed localized to injection site. This mouse fully recovered with no adverse effects. Please click here to view a larger version of this figure.
Discussion
The pEX100T-Not1 plasmid is an efficient mediator of sequential genomic deletions that are marker-free and in-frame. When engineering bacterial strains for attenuated virulence, deletion of entire gene sequences rather than generating point mutations decreases the likelihood of reversion to a virulent phenotype. Additionally, each pathogenicity gene deletion attenuates the pathogen further, reinforcing the stability of the attenuation.
This method can also be used to generate genomic modifications other than deletions, such as point mutations and insertions, simply by modifying the design of the plasmid insert. These types of modifications may be more useful than entire gene deletions for engineering bacteria with modified metabolism, for example. Sequential genomic modification has significant potential for generating designer bacterial strains to suit specific purposes in research and industry. Other methods of generating desired marker-free genomic modifications in bacteria have been described15,16,17,18. As with all genome-editing methods, attempted modifications to essential genomic regions may be lethal, and thus unsuccessful. In these cases, identification of different genetic modifications or other candidate genes is required to generate the bacterial strain of interest.
Given the numerous replication events and passages of each colony throughout this protocol, unintended changes to the genome will occur to the generated strain. The exact genomic changes can be identified through whole-genome sequencing. However, the impact of these changes is harder to determine. When engineering bacteria for a specific purpose, genomic changes that do not negatively affect the growth of the organism or the targeted pathway(s) are tolerable. Depending on the strain being generated, it may be possible to identify a "readout" to ensure that the strain is still useful for its intended purpose. For example, with PGN5, the goal was to create an attenuated strain that retained the ability to produce large amounts of alginate. After deletion of five pathogenicity genes, the amount and composition of alginate produced by PGN5 was measured and determined to be comparable to other alginate-producing strains. Thus, alginate production was unaffected by the five gene deletions, nor by the unintended genomic changes that occurred during the development of PGN5.
A model of intraperitoneal mouse injection was used to determine whether an engineered strain was attenuated compared to the parent strain and E. coli BL21, a strain approved by the FDA for production of biopharmaceuticals. The most important steps taken during this animal testing procedure were preparation and validation of frozen bacterial stocks. Preparation and use of frozen bacterial cultures to inject mice is preferable to using continuous culture, as it reduces the number mutations that naturally occur in bacterial populations19. Additionally, frozen cultures should remain viable for years. Viable plate counts showed no significant difference between the CFU/mL directly after stocks were prepared and three months after preparation. The use of multiple validation steps throughout this procedure ensured that the method was reproducible, and the results were not skewed by contaminating bacteria. Additionally, with the number of precautionary steps taken to ensure reproducibility, fewer animals were needed. Using a bacterial strain that is FDA-approved for biopharmaceutical production as the control (such as E. coli strain BL21), this method could be used to test the attenuation of other genetically engineered strains of P. aeruginosa, or other species of bacteria.
Using bioluminescence as a marker provides additional validation of the bacterial strains injected, as the marker can be visualized at the injection site. Insertion of the bioluminescence marker into the bacterial chromosome is required for bioluminescence imaging but may not be possible if working with incompatible strains/species. However, marking strains with bioluminescence is not required to test for attenuation. The strains tested in this study were marked with bioluminescence, which allowed for visualization of localization differences between strains throughout the course of the infection. We observed that the pathogenic strain disseminated through the body of the mouse, but the non-pathogenic strain remained at the site of injection. While this experiment only tested two very closely related strains of P. aeruginosa, it suggests that bacterial dissemination is linked to virulence, at least in P. aeruginosa. Thus, this procedure of labeling with bioluminescence to visualize the progression of the infection could be used in the future to quickly evaluate the attenuation of engineered strains of bacteria.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by the National Institutes of Health (NIH) grants R44GM113545 and P20GM103434.
Materials
| 0.2 mL tubes with flat caps | ThermoScientific | AB-0620 | via Fisher Scientific |
| 1 mL Syringe | BD | 22-253-260 | via Fisher Scientific |
| 1.5 mL disposable polystyrene cuvette | Fisher Scientific | 14955127 | |
| 1.5 mL Microcentrifuge Tubes | Fisher Scientific | 05-408-129 | |
| 2.0 mL Cryogenic Vials | Corning | 430659 | via Fisher Scientific |
| 27G needle | BD | 14-821-13B | via Fisher Scientific |
| 50 mL tubes | Fisher Scientific | 05-539-13 | via Fisher Scientific |
| Accu block Digital Dry Bath | Labnet | NC0205808 | via Fisher Scientific |
| Benchtop Centrifuge 5804R | Eppendorf | 04-987-372 | via Fisher Scientific |
| Benchtop Microcentrifuge | Sorvall | 75-003-287 | via Fisher Scientific |
| Cabinet Incubator | VWR | 1540 | |
| Carbenicillin disodium salt | Fisher Scientific | BP2648250 | |
| Culture Test Tube, Polystyrene | Fisher Scientific | 14-956-6D | via Fisher Scientific |
| Diposable Inoculation Loops | Fisher Scientific | 22-363-597 | |
| Dneasy UltraClean Microbial Kit (50) | Qiagen | 12224-50 | or preferred method/vendor |
| E.Z.N.A. Cycle Pure Kit (50) | Omega bio-tek | D6493-01 | or preferred method/vendor |
| EcoRI-HF, restriction endonuclease | New England BioLabs | R3101L | |
| Electroporation Cuvettes | Bulldog Bio | NC0492929 | via Fisher Scientific |
| FastLink II DNA Ligation Kit | Epicentre Technologies | LK6201H | via Fisher Scientific |
| Gentamycin Sulfate | Fisher Scientific | BP918-1 | |
| Glycerol | Fisher Scientific | BP229-4 | |
| GoTaq G2 Colorless Master Mix | Promega | M7833 | via Fisher Scientific |
| Isothesia Isoflurane | Henry Schein Animal Health | 29405 | |
| IVIS Lumina XRMS Series III In Vivo Imaging System | Perkins and Elmer | CLS136340 | |
| Kanamycin monosulfate | Fisher Scientific | BP906-5 | |
| LE agarose | Genemate | 3120-500 | via Fisher Scientific |
| Luria Broth | Difco | 240230 | via Fisher Scientific |
| MicroPulser Electroporator | BioRad | 1652100 | |
| Noble agar, ultrapure | Affymetris/USB | AAJ10907A1 | via Fisher Scientific |
| NotI-HF, restriction endonuclease | New England BioLabs | R3189 | |
| One Shot TOP10 Electrocomp E. coli | Invitrogen | C404052 | via Fisher Scientific |
| Phosphate buffered saline powder | Sigma | P3813-10PAK | Sigma-Aldrich |
| Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ | |
| Pseudomonas isolation agar | Difco | 292710 | via Fisher Scientific |
| Pseudomonas isolation broth | Alpha Biosciences | P16-115 | Custom made batch |
| QIAprep Spin Miniprep Kit (250) | Qiagen | 27106 | or preferred method/vendor |
| Shaking Incubator | New Brunswick Scientific | Innova 4080 | shake at 200 rpm |
| SimpliAmp Thermal Cycler | Applied Biosystems | A24811 | |
| Skim Milk | Difco | DF0032-17-3 | via Fisher Scientific |
| Small Plates (100 O.D. x 10 mm) | Fisher Scientific | FB0875713 | |
| SmartSpec Plus Spectrophotometer | Bio-Rad | 170-2525 | or preferred method/vendor |
| Sucrose | Fisher Scientific | S5-500 | |
| Toothpicks, round | Diamond | Any brand of toothpicks, autoclaved | |
| TOPO TA Cloning Kit, for seqeuncing | Invitrogen | 45-0030 | |
| XAF-8 Anesthesia System Filters | Perkins and Elmer | 118999 | |
| XGI 8 Gas Anesthesia System | Caliper Life Sciences/Xenogen |
References
- Gellatly, S. L., Hancock, R. E. Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathogens and Disease. 67 (3), 159-173 (2013).
- Valentine, M. E., et al. Generation of a highly attenuated strain of Pseudomonas aeruginosa for commercial production of alginate. Microbial Biotechnology. , (2019).
- Schweizer, H. P., Hoang, T. T. An improved system for gene replacement and xylE fusion analysis in Pseudomonas aeruginosa. Gene. 158 (1), 15-22 (1995).
- Damron, F. H., Qiu, D., Yu, H. D. The Pseudomonas aeruginosa sensor kinase KinB negatively controls alginate production through AlgW-dependent MucA proteolysis. Journal of Bacteriology. 191 (7), 2285-2295 (2009).
- Yu, H., Boucher, J. C., Hibler, N. S., Deretic, V. Virulence properties of Pseudomonas aeruginosa lacking the extreme-stress sigma factor AlgU (sigmaE). Infection and Immunity. 64 (7), 2774-2781 (1996).
- Baeshen, M. N., et al. Production of Biopharmaceuticals in E. coli: Current Scenario and Future Perspectives. Journal of Microbiology and Biotechnology. 25 (7), 953-962 (2015).
- Marisch, K., Bayer, K., Cserjan-Puschmann, M., Luchner, M., Striedner, G. Evaluation of three industrial Escherichia coli strains in fed-batch cultivations during high-level SOD protein production. Microbial Cell Factories. 12, 58 (2013).
- Ferrer-Miralles, N., Domingo-Espin, J., Corchero, J. L., Vazquez, E., Villaverde, A. Microbial factories for recombinant pharmaceuticals. Microbial Cell Factories. 8, 17 (2009).
- Horton, R. M., Cai, Z. L., Ho, S. N., Pease, L. R. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques. 8 (5), 528-535 (1990).
- Horton, R. M., Hunt, H. D., Ho, S. N., Pullen, J. K., Pease, L. R. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 77 (1), 61-68 (1989).
- Sambrook, J., Fritsch, E. F., Maniatis, T. . Molecular cloning: a laboratory manual. , (1989).
- Figurski, D. H., Helinski, D. R. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proceedings of the National Academy of Sciences of the United States of America. 76 (4), 1648-1652 (1979).
- Choi, K. H., Schweizer, H. P. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nature Protocols. 1 (1), 153-161 (2006).
- Choi, K. H., Kumar, A., Schweizer, H. P. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. Journal of Microbiological Methods. 64 (3), 391-397 (2006).
- Liang, R., Liu, J. Scarless and sequential gene modification in Pseudomonas using PCR product flanked by short homology regions. BMC Microbiology. 10, 209 (2010).
- Martinez-Garcia, E., de Lorenzo, V. Engineering multiple genomic deletions in Gram-negative bacteria: analysis of the multi-resistant antibiotic profile of Pseudomonas putida KT2440. Environmental Microbiology. 13 (10), 2702-2716 (2011).
- Song, C. W., Lee, S. Y. Rapid one-step inactivation of single or multiple genes in Escherichia coli. Biotechnology Journal. 8 (7), 776-784 (2013).
- Yan, M. Y., et al. CRISPR-Cas12a-Assisted Recombineering in Bacteria. Applied and Environmental Microbiology. 83 (17), (2017).
- Prakash, O., Nimonkar, Y., Shouche, Y. S. Practice and prospects of microbial preservation. FEMS Microbiology Letters. 339 (1), 1-9 (2013).
