This method is optimized for extracting cereal seed samples containing significant amounts of contaminating starch or sugars. It is designed to extract total RNA from 24 seed samples per day. It should be conducted continuously in one day, but optional stop and pause points are identified throughout the protocol. Alternatively, the reader can use their preferred RNA extraction kits or manual chemical extraction method. However, based on our previous experience, commercially available plant RNA extraction kits are not appropriate for seeds due to significant amounts of starch, proteins, sugar and/or lipid contamination. In the described method, a chemical extraction of crude RNA is followed by column purification using a commercial RNA extraction kit. This typically provides RNA with higher quality and yield. Figure 1 shows the result of a BioAnalyzer run to test the quality of RNA extraction using the method described here. Results are presented for barley leaf (Samples 1 and 2 representing low starch content samples) and barley seeds (Samples 3 and 4 representing high starch content samples). The RNA integrity (RIN) value for all samples is 10.
Figure 1 shows a representative gel of the purified RNA extract, where the quality was tested using a Bioanalyzer. Samples 1 and 2 are typical results for low starch content samples such as barley leaves, where additional rRNA bands from chloroplasts are evident. Samples 3 and 4 are representative results for high starch content samples such as barley seeds, showing 18S and 28S rRNA. Please note that no automated RIN value can be calculated for green plant tissues such as leaf samples, due to chloroplast rRNA. However, the integrity and high quality can be visually ascertained by to the absence of any degradation products, which typically appears as low molecular smear. The RIN value for high-starch seeds samples such as barley seeds is typically 10 using the protocol described in this paper.
In addition, we also present here a representative data of a time course experiment of two elite barley inbred lines (Sofiara and Victoriana) used for analysis of malting quality19. During the malting process, starch is converted into sugar. Therefore, the samples represent tissues with varying proportions of starch and sugar contents. As the industrial malting process is similar to the germination process, the transcriptomes of two barley lines, differing in their malting quality, were analyzed. RNAs were extracted from germinating seeds at 2, 24, 48, 72, 120, 144 and 196 h after imbibition in biological triplicates. The RNA preparation and hybridization to the customized barley microarray chip were performed as described above. Figure 2 indicates the normalized grid read out from the microarray hybridization given in the quality control (QC) report (Figure 3) and the histogram plot for detected signals. The grid gives the example of derived signals from each corner of the chip, including background and spike-in read out dots used for calibration. The histogram indicates the deviation of detectable dots with respective signal intensities. A successful hybridization gives a broad Gaussian-shaped curve with only minor outliers as shown in the figure. Failed hybridizations can result in a strong shift towards one side ("green monsters").
Lastly, a representative result of the acceptable values is shown in column 4 of Figure 3. To indicate the reliability of the performed experiments, the results are further evaluated using the GeneSpring software. The collected data are presented as principal component analysis (PCA). The PCA integrates the values of selected dots (genes) as vector. The number of evaluated dots that are used can vary from several hundred to the entire chip and is dependent on the software used. Each chip (sample) results in one value (vector) that resulted from the integrated signal intensities for the analyzed dots. Therefore, the relative position in the graph (PCA) indicates the similarity of the samples to each other. The closer the samples are, the more similar they are. Technical replicates should be closer together than biological ones, and biological replicates of a sample should cluster closer together, than samples from different time points, tissues or conditions.
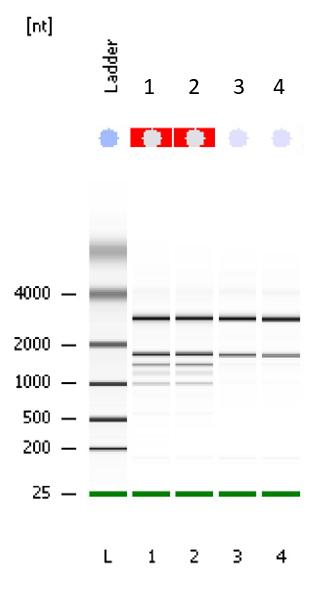
Figure 1: Electrophoresis file run summary obtained after checking the quality of RNA with Bioanalyzer. Samples 1 and 2 are barley leaf tissue as indicated by additional ribosomal bands from chloroplasts. Samples 3 and 4 are barley seed tissue with 18S and 28S rRNA bands shown. A RIN factor is not always calculated for green tissues such as leaf but according to the gel, the quality of RNA is very good. The RIN value for samples 3 and 4 is 10. Please click here to view a larger version of this figure.

Figure 2: QC Report from successful barley microarray hybridization. A + indicates detected signals on the grid from all corners of the chip. The histogram shows the number of signals categorized according to signal intensity (fluorescence) as logarithm after background subtraction. Please click here to view a larger version of this figure.
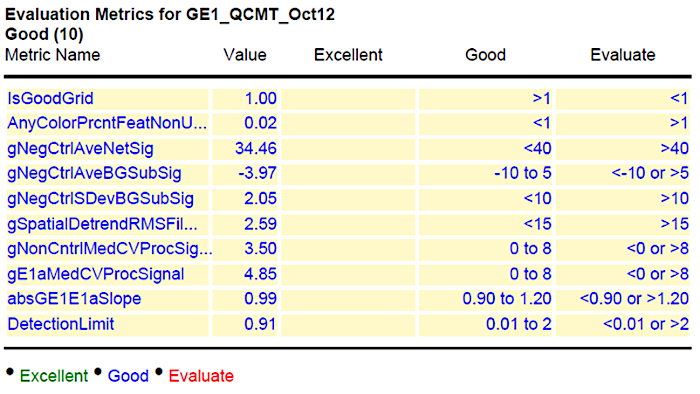
Figure 3: Summary of quality control (QC) after hybridization and scanning. The values for the hybridized slide are given in column 2 (value) and the range of acceptable values is shown in column 4. Please click here to view a larger version of this figure.
| Wash Chamber Assembly | Content and Label | Purpose |
| Dish 1 | Empty, leave on lab bench until next day | Fill with Wash Buffer 1 the next day, used to disassemble the microarray slides |
| Dish 2 | Add one microarray slide rack and a small magnetic stir bar; label with "Wash Buffer 1", leave on lab bench until next day | Used to wash the microarray slides with Wash Buffer 1 the next day |
| Dish 3 | Add one small magnetic stir bar, label with "Wash Buffer 2" and place in a 37 °C mini incubator. | Used to wash the microarray slides with Wash Buffer 2 the next day inside the 37 °C mini incubator |
Table 1: Preparation of the wash chamber assemblies.
| Steps | Dish | Wash Buffer | Temperature | Time |
| Disassembly | 1 | 1 | Ambient | As fast as possible |
| (Step 4.13) | ||||
| First wash | 2 | 1 | Ambient | 1 min |
| (Step 4.14) | ||||
| Second wash | 3 | 2 | 37 °C | 1 min |
| (Step 4.15) |
Table 2: Incubation temperature and time for wash chamber assemblies.
