Single-molecule Förster resonance energy transfer (smFRET) is a technique that measures the FRET efficiency between two dyes-a donor and an acceptor-at the level of individual molecules. FRET is a photophysical process arising from the overlapping energy spectra of two dyes: the donor is excited by light of a specific wavelength and transfers energy non-radiatively to the acceptor, resulting in emission from the acceptor. The efficiency of this transfer is inversely proportional to the sixth power of the distance between the two dyes, so the transfer efficiency varies with distance1. Thus, this FRET efficiency can be used to determine spatial information about the molecule(s)2 to which the dyes are attached, within a range of 3-10 nm. This scale, and the fact that changes in FRET efficiency are sensitive to Angstrom molecular movements3, makes the technique well suited to investigating structural information about biomolecules-such as nucleic acids and proteins-without the complications of ensemble averaging4,5,6. While changes in relative FRET efficiencies can be used to monitor biomolecular interactions and conformational dynamics, shedding light on key cellular processes such as protein (un)folding, transcription, and DNA replication and repair, absolute FRET efficiencies have been used to determine precise distances for biomolecular structure determination7,8,9,10,11, overcoming the need for crystallization or freezing as is required for some other structural methods4,12.
smFRET experiments most commonly take two forms, confocal or total internal reflection fluorescence (TIRF) microscopy. Between both approaches the molecular dynamics of biomolecules can typically be investigated on timescales from pico- to millisecond (confocal, freely diffusing molecules) up to millisecond to hours (TIRF, surface immobilized molecules). This is due to the different setups involved in each technique. In TIRF microscopy, molecules are immobilized on the surface of a slide and excited by an evanescent wave (Figure 1A). Here, however, the focus is on confocal microscopy as this is the format of the smfBox. In confocal microscopy, molecules are not immobilized and instead freely diffuse via Brownian motion through the confocal volume (~1 fL), formed by focusing a laser beam through a high numerical aperture lens into a spot at some designated depth within the solution (Figure 1B). The resulting emission is focused back through the same aperture and filtered through a dichroic mirror (Figure 1C for full schematic). It is then focused through a pinhole in order to remove any out-of-focus light and onto an avalanche photodiode (APD). When the APD detects a photon, it outputs a TTL pulse, the timing of which can be recorded with up to picosecond resolution. The observation time of these freely diffusing molecules within the vicinity of the confocal volume is commonly within the order of milliseconds.
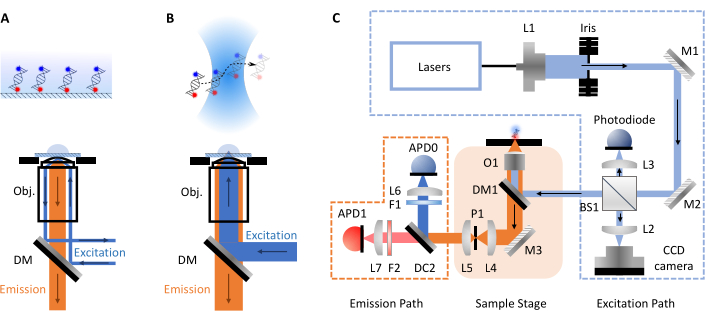
Figure 1: Schematics showing principles of microscopy and the smfBox setup. (A) Total Internal Reflection Fluorescence (TIRF) Microscopy principle: excitation light is directed into the edge of the objective (Obj.) and undergoes total internal reflection at the coverslip-buffer interface generating an exponentially decaying evanescence field to excite surface attached molecules. (B) Confocal Microscopy: Freely diffusing molecules are excited by a near diffraction-limited spot focused into the sample. (C) The smfBox setup used in this protocol, showing all key components: avalanche photodiodes (APD), beam splitter (BS), dichroic mirrors (DM), filters (F), mirrors (M), objective (O) and pinhole (P). Please click here to view a larger version of this figure.
More recently, smFRET techniques incorporated two color excitation, where lasers matching the donor and acceptor excitation wavelengths are alternated5. This can be done in one of two ways, the first by modulating continuous wave lasers on the KHz timescale, which is known as alternating laser excitation (ALEX)13,14. The second method interleaves fast pulses on the MHz timescale; this is nanosecond-ALEX15 or pulsed interleaved excitation (PIE)16. In all these approaches, information from the acceptor laser leads to calculation of the so-called stoichiometry, which can discriminate between molecules with a low FRET efficiency and those lacking an acceptor (either through incomplete labeling or photobleaching). Using PIE/ns-ALEX additionally gives access to fluorescent lifetimes on the single-molecule level, and anisotropies can be measured when coupled with polarizing optics. This combination of measurements is known as multiparameter fluorescence detection (MFD)9.
Despite the many advantages of smFRET, it is not widely used outside of specialist labs due to the high costs of commercial instruments and a lack of simple, self-build alternatives. A growing trend towards development of low-cost opensource microscopy is taking place and other platforms have recently emerged, including Planktonscope17, OpenFlexure Microscope18, Flexiscope19, miCube20, liteTIRF21, and Squid22. Herein the study describes the protocol for using the smfBox, a recently developed cost-effective confocal set-up capable of measuring the FRET efficiency between two dyes on freely diffusing single molecules. Detailed build instructions and all the necessary operational software are freely available at: https://craggslab.github.io/smfBox/23. The optical arrangement of the smfBox is assembled from readily available components purchased from affordable and widely-accessible manufacturers, while the microscope body (responsible for the majority of the expense in a standard confocal set-up) has been replaced by a custom light-tight anodized-aluminium box (allowing measurements to be made under ambient light conditions). This box houses key optical components, including the excitation dichroic, objective, and pinhole, and a mechanical laser interlock, enabling its safe operation as a Class I laser product (see Figure 1C for a full schematic). The smfBox uses ALEX to validate the dye stoichiometry and to determine accurate FRET correction factors. It is operated using custom-written, open-source software (smOTTER), which controls all aspects of the data acquisition and outputs the data in the open-source photon-HDF5 format24, compatible with many third-party analysis tools. The smfBox and the acquisition and data analysis protocols were recently tested against >20 other instruments (both confocal and TIRF) in a multi-lab blind study25. The FRET efficiencies obtained were in excellent agreement with all the other instruments, despite the smfBox costing only a fraction of the price of commercially available setups.
Here, a step-by-step protocol is outlined for acquiring and analyzing accurate, absolute FRET efficiencies on freely diffusing DNA duplexes using the smfBox, all the way from switch on, through alignment and focusing, to data collection and analysis. The samples used here are three duplex DNAs (exhibiting high-, mid- and low-FRET efficiencies, see Table 1) that were assessed in the world-wide blind study25; however, the method is adaptable to many molecular systems, including proteins and other nucleic acids. The hope is that such a detailed protocol, along with the already existing build instructions for the smfBox23, will help to make this powerful technique even more accessible to a wide range of labs.
The protocol necessitates critical assessment of experimental conditions during setup (see protocol step 4.8). The first results acquired which determine success or failure of the experiment are achieved at this stage. A positive result would be to have between five and one bursts per second (see Figure 2B,C). A negative result would be having too many (Figure 2A) or too few bursts (Figure 2D) within that time frame. It remains possible at this stage to rectify these errors: a sample with too high a concentration needs simply to be diluted; if the concentration is too low, however, a new sample may need to be prepared (the determinant being whether it remains possible at this low concentration to collect data in a reasonable time frame).
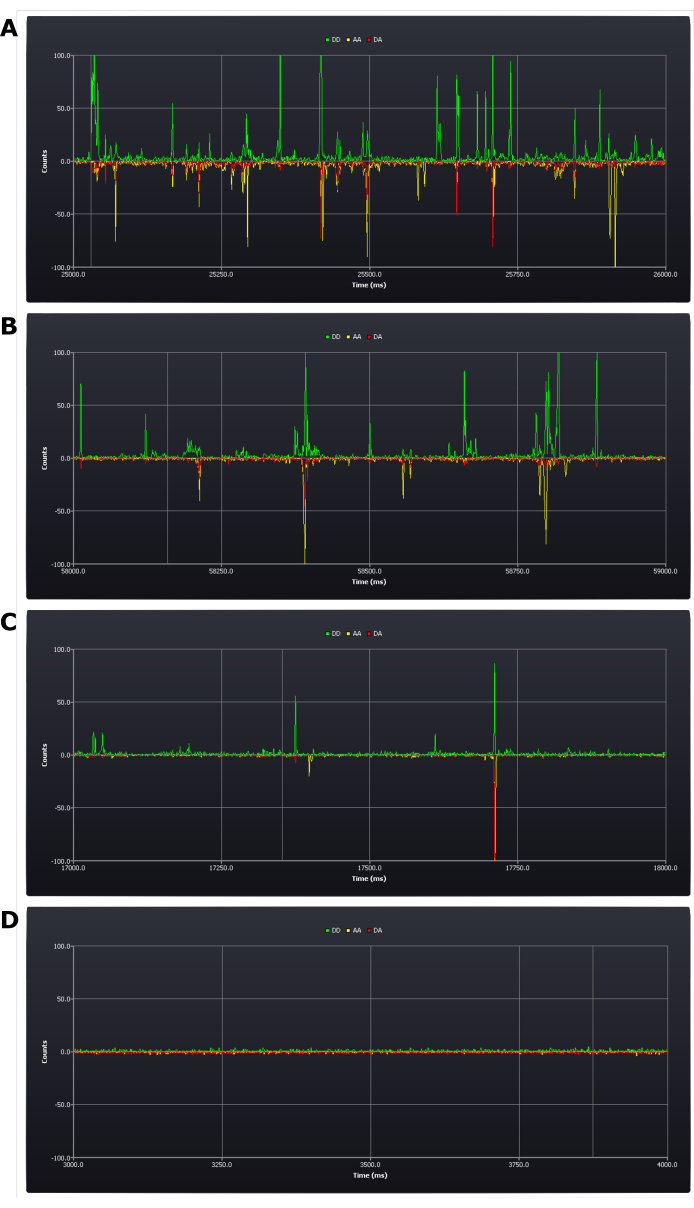
Figure 2: Screen shots from live trace during experimental setup showing different concentrations of doubly labeled duplex DNA samples. (A) too high, (B) upper acceptable limit, (C) target concentration, (D) too low. Photon counts (1 ms bins) are shown in the three detection channels; donor emission after donor excitation (DD), acceptor emission after donor excitation (DA), and acceptor emission after acceptor excitation (AA). Please click here to view a larger version of this figure.
A static single-species system would typically require 30 to 60 min of measurement to obtain the necessary ~1,000 bursts needed for robust data analysis. The length of time and number of bursts required will increase with multiple species or dynamic systems. Following data collection and analysis using the protocol figures are exported from the Jupyter notebooks. The alternation plot (Figure 3A) should match the ALEX period of the experimental setup. The time trace (Figure 3B) is used to qualitatively assess that the sample concentration is reasonable. The background plot (Figure 3C) shows the distribution of inter-photon delay periods with a linear fit to the longer times to estimate the background rate26. The background trace (Figure 3D) can identify if there were changes in the sample over the duration of the experiment; primarily this would be due to evaporation during longer acquisition times. ES histograms are generated for all photons (Figure 3E) and doubly labeled species (Figure 3F). Finally, a 1D E histogram (Figure 3G) is generated with gaussian fitting of the burst data.
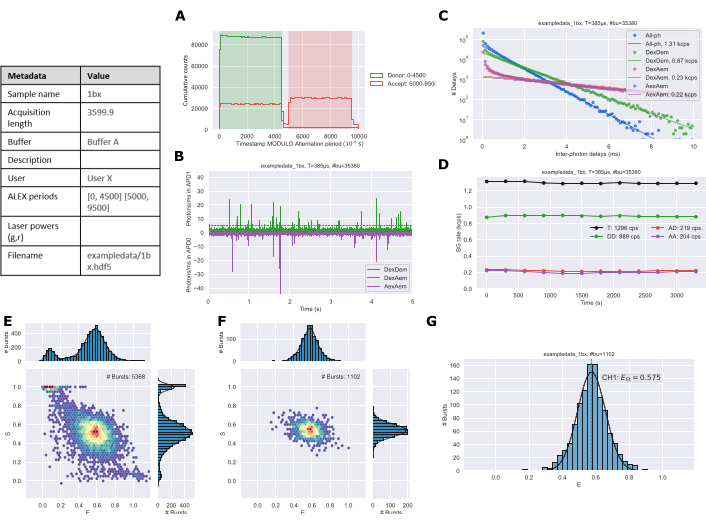
Figure 3: Example figure output of analyzed data generated by the Jupyter Notebooks. (A) Alternation plot, (B) Time trace, (C) Background determination, (D) Background rates, (E) All photon ES histogram, (F) Dual channel ES histogram, and (G) 1D E histogram. Please click here to view a larger version of this figure.
| Name | Sequence |
| 1a | 5’- GAG CTG AAA GTG TCG AGT TTG TTT GAG TGT TTG TCT GG – 3’ |
| 3’- CTC GAC TTT CAC AGC TCA AAC AAA CTC ACA AAC AGA CC – 5’ | |
| 1b | 5’- GAG CTG AAA GTG TCG AGT TTG TTT GAG TGT TTG TCT GG – 3’ |
| 3’- CTC GAC TTT CAC AGC TCA AAC AAA CTC ACA AAC AGA CC – 5’ | |
| 1c | 5’- GAG CTG AAA GTG TCG AGT TTG TTT GAG TGT TTG TCT GG – 3’ |
| 3’- CTC GAC TTT CAC AGC TCA AAC AAA CTC ACA AAC AGA CC – 5’ |
Table 1: DNA sequences used in the protocol. Nucleotides are highlighted in blue and red representing C2 amino modified thymine residues labeled with Atto-550 and Atto-647N, respectively.
Correction Factor Finder Alpha-Delta: Please click here to download this File.
Correction Factor Finder Gamma-Beta: Please click here to download this File.
FRET Analysis 1.4 Corrected: Please click here to download this File.
FRET Analysis 1.4 Uncorrected: Please click here to download this File.
