Carr-Purcell Meiboom-Gill (CPMG) relaxation dispersion (RD) experiments are used on a routine base to characterize conformational equilibria occurring on the µs-ms timescale by solution NMR spectroscopy1,2,3,4,5. Compared to other methods for investigation of conformational dynamics, CPMG techniques are relatively easy to implement on modern NMR spectrometers, do not require specialized sample preparation steps (i.e., crystallization, sample freezing or alignment, and/or covalent conjugation with a fluorescent or paramagnetic tag), and provide a comprehensive characterization of conformational equilibria returning structural, kinetic, and thermodynamic information on exchange processes. In order for a CPMG experiment to report on a conformational equilibrium, two conditions must apply: (i) the observed NMR spins must possess different chemical shifts in the states undergoing conformational exchange (microstates) and (ii) the exchange has to occur at a time scale ranging from ~50 µs to ~10 ms. Under these conditions, the observed transverse relaxation rate ( ) is the sum of the intrinsic R2 (the R2 measured in the absence of µs-ms dynamics,
) is the sum of the intrinsic R2 (the R2 measured in the absence of µs-ms dynamics,  ) and the exchange contribution to the transverse relaxation (Rex). The Rex contribution to R2obs can be progressively quenched by reducing the spacing between the 180° pulses constituting the CPMG block of the pulse sequence, and the resulting RD curves can be modeled using the Bloch-McConnell theory to obtain the chemical shift difference among microstates, the fractional population of each microstate, and the rates of exchange among microstates (Figure 1)1,2,3.
) and the exchange contribution to the transverse relaxation (Rex). The Rex contribution to R2obs can be progressively quenched by reducing the spacing between the 180° pulses constituting the CPMG block of the pulse sequence, and the resulting RD curves can be modeled using the Bloch-McConnell theory to obtain the chemical shift difference among microstates, the fractional population of each microstate, and the rates of exchange among microstates (Figure 1)1,2,3.
Several different pulse sequences and analysis protocols have been reported in the literature for 15N CPMG experiments. Herein, the protocol implemented in the laboratory is described. In particular, the crucial steps for preparation of the NMR sample, set up and acquisition of the NMR experiments, and processing and analysis of the NMR data will be introduced (Figure 2). To facilitate transfer of the protocol to other laboratories, the pulse program, processing and analysis scripts, and one example dataset are provided as Supplemental Files and are available for download at (https://group.chem.iastate.edu/Venditti/downloads.html). The provided pulse sequence incorporates a four-step phase cycle in the CPMG block for suppression of offset-dependent artifacts6 and it is coded for acquisition of several interleaved experiments. These interleaved experiments have an identical relaxation period but different numbers of refocusing pulses in order to achieve different CPMG fields7. It is also important to notice that the described pulse program measures the 15N R2 of the TROSY component of the NMR signal8. Overall, the protocol has been successfully applied for the characterization of conformational exchange in medium and large-sized proteins4,5,9,10. For smaller systems (<20 kDa), the use of an Heteronuclear Single Quantum Coherence (HSQC)-based pulse sequence11,12 is advisable.
1. Preparation of the NMR sample
- Express and purify a 2H,15N-labled sample of the protein of interest.
NOTE: While a 15N-labeled protein sample can be used for acquisition of the CPMG RD experiment, perdeuteration (where possible) dramatically increases the quality of the obtained data. Protocols for the production of perdeuterated proteins are available in the literature13. - Buffer exchange the purified protein sample into a degassed NMR buffer.
- Transfer the NMR sample into the NMR tube.
NOTE: The concentration of the NMR sample needs to be carefully optimized in order to maximize the signal-to-noise ratio and minimize the occurrence of protein-protein interactions. In general, the typical concentration range for CPMG experiments is 0.1-1.0 mM.
2. First time set-up of the NMR experiment
- Download and unzip the Supplemental Files.
- Copy the files bits.vv and trosy_15N_cpmg.vv (located the in the folder named pulseprogram) into the pulse program directory (<path>/exp/stan/nmr/lists/pp/user).
NOTE: Make sure the path to bits.vv listed on the first line of the trosy_15N_cpmg.vv file is correct. - Open acquisition software and copy a previously run 1H-15N HSQC experiment into a new experiment using the command edc.
- Using the command pulprog, load the pulse program trosy_15N_cpmg.vv into the newly created experiment.
- Set up the CPMG experiment using the instructions provided at the end of the pulse program file (trosy_15N_cpmg.vv).
3. Routine set-up of the NMR experiment
- Introduce the sample in the magnet and perform all the basic steps for acquisition of any NMR experiment: lock and shim the sample; match and tune the 1H and 15N channels.
- Set p1 and p7 to the duration of the 1H and 15N hard 90° pulses, respectively.
NOTE: For optimal results, it is important to calibrate the 15N 90° pulse with great care. Calibration is usually accomplished using a 100 mM sample of 15N-labeled urea in DMSO as described in the spectrometer manual. In addition, it is possible to double-check the calibration directly on the working NMR sample by acquiring the first increment of a 1H-15N HSQC spectrum in which the 15N 90° pulse of the first INEPT block is switched to a 180° pulse. If the calibration is correct, a null should be obtained. - In the acquisition window, set the center and spectral width for the 1H and 15N dimensions.
NOTE: Center the 1H spectrum at the frequency of the water signal for optimal water suppression. - Set the relaxation delay (d30) equal to 0.7 T2, where T2 is the expected 15N transverse relaxation time of your protein.
NOTE: The value of d30 can be optimized empirically to obtain the best results. - Use the command vclist to create a list of integer numbers corresponding to the parameter n in Figure 1A and Figure 1B. Ensure that each entry in the list corresponds to a different CPMG field (νcpmg) according to νcpmg = 4 x n / d30. Make sure that the first number in the list is 0 (this corresponds to the reference experiment for which the CPMG block is skipped and d30 = 0 s) and not to use numbers larger than 1,000 x d30 / 4. Numbers larger than this threshold result in νcpmg > 1 kHz and might damage the probe.
- Set l8 to the number of entries in the vclist, l3 to the number of real points for the indirect dimension (usually a range of 64-200 is set for l3), and 1 td equal to l8 x l3 x 2.
NOTE: Perform the steps 3.7-3.11 to optimize the water suppression. - Set the receiver gain (rg) to 1; open the pulse program file (edcpul), go to line 91, remove the semicolon preceding goto 999, and save the file.
- Using the command gs adjust the parameters spdb0 (or spdw0) in order to minimize the intensity of the FID signal.
- Reintroduce the semicolon at line 91 of the pulse program file and save the file.
- Repeat the steps 3.7-3.9 to optimize spdb11 (line 168 of the pulse program file), spdb2 (line 179 of the pulse program file), and pldb2 (line 184 of the pulse program file).
- Run the command rga to optimize the receiver gain.
- Set the number of scans (ns) to a multiple of 8.
- Run the command zg to start the experiment.
4. Processing and analysis of the NMR data
- Copy the folder named Process (Supplemental Files) into the directory containing the ser file.
- Use the files sep_fid.com and ft2D.com to process the NMR data.
NOTE: Instruction as to how to edit the processing scripts is provided within the sep_fid.com and ft2D.com files. - Open the ucsf files contained in the directory CPMG_Sparky_files in Sparky.
NOTE: The ucsf files are created by the processing scripts. There is one ucsf file for each entry in the vclist. The first ucsf file (test_1.ucsf) contains the reference experiment. The subsequent ucsf files (test_2.ucsf, test_3.ucsf,… test_n.ucsf) are ordered from the lowest to the largest value of νcpmg. - Pick the NMR cross-peaks on the reference NMR spectrum (test_1.ucsf).
- Select all the picked cross-peaks using the Sparky command pa.
- Run the Sparky command rh. This command opens a dialogue window. Select the option heights at the same position in each spectrum. Click on Setup and check all the NMR spectra. Click on Update and save the output file in the working directory. The output file contains the matrix of the signal intensities over all opened NMR spectra.
NOTE: More accurate protocols that use lineshape fitting for recovering intensities from interleaved pseudo-3D experiments have been described in the literature14. Freely available software for lineshape fitting is available at https://pint-nmr.github.io/PINT/ (PINT), https://www.ucl.ac.uk/hansen-lab/fuda/ (FUDA), and within NMRPipe (nLinLS module) and SPARKY (it module). - For each CPMG field, convert the signal intensities to R2 rates using the formula
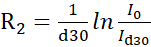 , where I0 and Id30 are the intensity of the reference (vc = 0) and relaxed (vc > 0) NMR spectra, respectively.
, where I0 and Id30 are the intensity of the reference (vc = 0) and relaxed (vc > 0) NMR spectra, respectively.
NOTE: In the Supplemental Files, a template of the spreadsheet file (R2_calc.xls) used to convert the signal intensities to R2 rates and to visualize the RD profiles is provided. - Read the noise level in the reference spectrum using the Sparky command st and propagate the error on the R2 rates.
NOTE: In the Supplemental Files, a template of the spreadsheet file (R2_calc.xls) used to propagate the error on the measured R2 rates is provided.
5. Fitting RD curves
- Copy the folder named RD_fitting (Supplemental Files) into the working directory.
- For each assigned NMR peak, estimate Rex by calculating the
 .
.  and
and  are the R2 rates measured at the lowest and highest νcpmg in the vclist.
are the R2 rates measured at the lowest and highest νcpmg in the vclist. - Visually inspect the RD curves with Rex larger than two times the estimated error and discard all the RD curves that are too noisy to be modeled accurately.
NOTE: Noise on Rex can be propagated from the errors on and . - Prepare an input file for the fitting script using all RD curves with Rex larger than two times the estimated error.
NOTE: Detailed instruction on the preparation of the input files are provided within the fitting scripts. Example input files are provided as Supplemental Files. Commonly, RD data are measured at two different static fields and fitted simultaneously. In this protocol, two different input files are required (Supplemental Files). - Fit the RD data using the scripts provided in Supplemental Files.
NOTE: Two different scripts are provided to perform a residue-based or a global fit of the RD curves, respectively. Both scripts fit the RD curves using a two-site exchange model and the Carver-Richards equation. More detailed instructions are provided within the fitting scripts. Additional software packages such as Chemex (https://github.com/gbouvignies/ChemEx) and CATIA (https://www.ucl.ac.uk/hansen-lab/catia/ ) are available to carry out data fitting using the Block-McConnell equations. - Test the reliability of the fitted parameters estimating the reduced χ2 as a function of pb and kex.
NOTE: The reduced χ2 is provided in the output file. pb and kex can be restrained to specific values using lower and upper bounds in the fitting procedures. In our scripts, the lower and upper bounds for pb are lb(2) and ub(2), respectively. The lower and upper bounds for kex are lb(3) and ub(3), respectively. - Estimate the error on the fitted parameters. This can be done by setting the value of MC_fac in the script to 1 and repeating the fitting multiple times (typically >20 repeats). The error on each parameter is estimated as the standard deviation of the distribution.
NOTE: Setting MC_fac to 1 generates a synthetic dataset in which a Gaussian distributed error (calculated based on the experimental error) is added to the experimental data.
The protocol described here results in acquisition of RD profiles for each peak in the 1H-15N TROSY spectrum (Figure 3A). From the acquired RD profiles, it is possible to estimate the exchange contribution to the 15N transverse relaxation of each backbone amide group (Figure 3A,3B). By plotting the Rex on the 3D structure of the protein under investigation, it is possible to identify the structural regions undergoing conformational exchange on the µs-ms time scale (Figure 3C). Modeling of the RD curves using the Carver-Richards equation returns thermodynamic and kinetic parameters on the exchange process, such as the fractional populations of the states in equilibrium and the rate of exchange among these states (Figure 1, Figure 3D). The temperature dependence of these thermodynamic and kinetic parameters (obtained by acquiring RD experiments at multiple experimental temperatures) can be modeled using the van't Hoff and Eyring equations, respectively, to obtain detailed information on the energetics of the conformational exchange (Figure 3E)9,10.

Figure 1: Overview of the CPMG RD experiment. (A) Schematic view of the CPMG block used for acquisition of RD data. The 180° pulses are shown as black rectangles. The operator  indicates the magnetization that enters and exits the CPMG block. The CPMG field is determined by the spacing between subsequent refocusing pulses (2τ). (B) In a two-time point measurement, the relaxation delay (during which the CPMG block is applied) is kept constant and the CPMG field is varied by varying the values of n (the number of times the CPMG block is applied during the relaxation delay period) and τ. (C) Schematic representation of a two-site equilibrium between conformations A and B. The exchange rate constant (kex) is the sum of the forward and reverse rate constants kab and kba, respectively). pa and pb (= 1 – pa) are the fractional populations of species A and B, respectively. Δωab is the chemical shift difference between conformations A and B. (D) Simulated RD curves for exchange processes that are in the range accessible by CPMG (top left), too fast to be detected by CPMG (top right), too slow to be detected by CPMG (bottom right), and with a Δωab too small to be detected by CPMG (bottom left). The pb value was set to 3%. Please click here to view a larger version of this figure.
indicates the magnetization that enters and exits the CPMG block. The CPMG field is determined by the spacing between subsequent refocusing pulses (2τ). (B) In a two-time point measurement, the relaxation delay (during which the CPMG block is applied) is kept constant and the CPMG field is varied by varying the values of n (the number of times the CPMG block is applied during the relaxation delay period) and τ. (C) Schematic representation of a two-site equilibrium between conformations A and B. The exchange rate constant (kex) is the sum of the forward and reverse rate constants kab and kba, respectively). pa and pb (= 1 – pa) are the fractional populations of species A and B, respectively. Δωab is the chemical shift difference between conformations A and B. (D) Simulated RD curves for exchange processes that are in the range accessible by CPMG (top left), too fast to be detected by CPMG (top right), too slow to be detected by CPMG (bottom right), and with a Δωab too small to be detected by CPMG (bottom left). The pb value was set to 3%. Please click here to view a larger version of this figure.
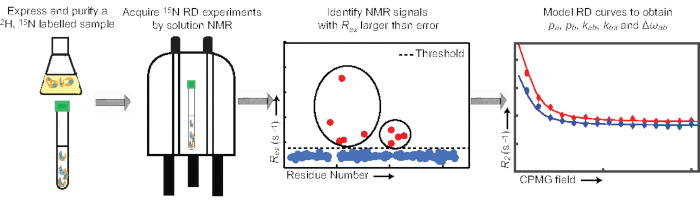
Figure 2: Main pipeline for acquisition and analysis of RD data. Schematic representation of the workflow described in the present protocol. Please click here to view a larger version of this figure.

Figure 3: µs-ms dynamics of EIC by CPMG RD experiments. (A) Example 15N RD profiles measured at 40 °C and 800 MHz for EIC using the protocol described here. The estimation of Rex is shown in red. (B) Rex values are plotted versus the residue index and (C) on the X-ray structure of the enzyme to identify residues undergoing conformational exchange. (D) NMR signals with Rex larger than the error are modeled (using the script provided in Supplemental Files) to obtain the kinetics (kab and kba) and thermodynamics (pb) of the equilibrium. (E) Acquisition of RD data at multiple temperatures returns information on the energetics of the conformational change. Please click here to view a larger version of this figure.
Supplemental Files. Please click here to download this File.
