Single-Cell Characterization of Calcium Influx and HIV-1 Infection using a Multiparameter Optofluidic Platform
Summary
Here, we present a protocol in which single cells are monitored for acute events and productive HIV-1 infection on a nanofluidic device. Imaging data define virus-host receptor interactions and signaling pathway dynamics. This is the first method for nanofluidic high-throughput longitudinal single-cell culture and imaging to study signaling kinetics and molecular interactions.
Abstract
HIV-1 causes a chronic infection that affects more than 37 million people worldwide. People living with human immunodeficiency virus (HIV) experience comorbidity related to chronic inflammation despite antiretroviral therapy. However, these inflammatory signaling has not been fully characterized. The role of early entry events on the activation of cellular signaling events and downstream gene expression has not been captured at the single-cell level. Here the authors describe a method that applies principles of live-cell fluorescence microscopy to an automated single-cell platform that cultures and images cells over user-customized time courses, allowing for high-throughput analysis of dynamic cellular processes. This assay can track single-cell live fluorescence microscopy of early events that immediately follow HIV-1 infection, notably the influx of calcium that accompanies exposure to the virus and the development of productive infection using a fluorescent reporter virus. MT-4 cells are loaded with a calcium-sensitive dye and cultured in isolated pens on a nanofluidic device. The cultured cells are infected with an HIV-1 reporter virus (HIV-1 NLCI). A fluorescence microscope positioned above the nanofluidic device measures calcium influx over an 8-min time course following acute HIV-1 exposure. HIV-1 productive infection is measured in those same cells over a 4-day interval. Imaging data from these time courses are analyzed to define virus-host receptor interactions and signaling pathway dynamics. The authors present an integrated, scalable alternative to traditional imaging methods using a novel optofluidic platform capable of single-cell sorting, culturing, imaging, and software automation. This assay can measure the kinetics of events under various conditions, including cell type, agonist, or antagonist effect, while measuring an array of parameters. This is the first established method for nanofluidic high-throughput longitudinal single-cell culture and imaging: This technique can be broadly adapted to study cellular signaling kinetics and dynamic molecular interactions.
Introduction
Chronic inflammation is a leading cause of HIV-associated early morbidities and mortality1,2,3. There are multiple mechanisms whereby HIV can activate inflammatory signaling, and recent evidence suggests a role for the P2X receptors in HIV entry which are calcium-gating adenosine triphosphate (ATP) receptors3,4,5,6,7,8,9,10. The P2X subtype of purinergic receptors (P2XR) may be important facilitators of this inflammation. However, the molecular mechanisms of HIV-P2XR interactions are largely unknown and may impact early and late HIV-1 viral life cycle steps. Defining the pathways and kinetics driving HIV-associated chronic inflammation is critical to advancing treatment options for people with HIV.
To assess whether HIV-1 directly agonizes P2X receptors, P2XR activation and HIV-1 infection must be measured in parallel. Assays of P2XR activity and HIV-1 infection have been independently established: Cellular calcium influx is an indicator of P2XR activation, and HIV-1 productive infection can be quantified by RNA abundance. Fluorescence detection of calcium influx is possible with the Fluo-4 calcium-sensitive dye, and HIV-1 infection can be visualized with the mCherry fluorescent reporter virus HIV-NLCI11,12,13,14,15.
Because these indicators of P2X activation (acute cellular calcium influx) and HIV-1 infection (HIV-1 RNA synthesis) occur on different timescales (minutes versus days), there lacks a high-throughput method that allows for paired analysis of P2XR activation and HIV-1 infection. Standard high-throughput experimental techniques, such as flow cytometry, allow for the population analysis but cannot assess the relationship between acute and longitudinal events in single cells. Alternatively, single-cell imaging with standard fluorescence microscopy is low-throughput. These experimental limitations present a need for novel, high-throughput techniques to measure associations between acute and longitudinal cellular events directly.
An optofluidic system described is a novel platform capable of single-cell sorting and isolation, culturing, imaging, and software automation16,17,18,19. This system presents an integrated, high-throughput alternative to the limitations of traditional imaging methods. The Beacon platform consists of a carbon dioxide (CO2) and temperature-controlled incubator that supports cells contained on a chip. The chip possesses photosensitive transistors that generate an electrical gradient in response to targeted light. This resulting dielectrophoretic force is used to move individual cells across the nanofluidic chip to the desired regions. Cells are sorted into pens on the chip, which provide a barrier to isolate individual cells physically. Continuous laminar flow of growth media throughout the chip prevents cell migration from the pens while allowing for small-particle diffusion of nutrients and experiment-specific reagents. A fluorescence microscope sits above the chip. Software automation is used to capture images of the chip at the user-specified time point.
All cell characterization was performed using an optofluidic system for single-cell selection and manipulation. This system consists of integrated mechanical, microfluidic, and optical components that enable single-cell manipulation, assay, culture, and imaging. Cells are loaded and cultured on the disposable nanofluidic device consisting of 3,500 individual chambers (pens), each capable of holding sub-nanoliter volume. Cells can be positioned within pens using light-induced dielectrophoretic "cages" and cultured under temperature- and CO2-controlled conditions. The microfluidics permit perfusion of media or buffers on the chip for cell culture or drug treatment. An actuated needle allows for the import and export of cells from incubated and shuttered well plates. The chip area can be imaged at 4x or 10x magnification in brightfield and fluorescent channels (including DAPI, FITC, TRed, or Cy5) to characterize cellular phenotypes or functional analysis. The entire system is automated using software that can be used for predesigned workflows or custom experiments.
Relationships between HIV-1 infection and P2XR have been studied, but a high-throughput procedure to directly characterize these interactions in parallel has not been reported. Here, the authors describe a methodology to study HIV-P2XR interactions through tracking acute calcium influx and subsequent HIV-1 productive infection at the single-cell level. Notably, this establishes a novel tool that allows for direct, high-throughput, longitudinal measurement of multiple targets in single cells.
Protocol
1. Preparation of cells for imaging
- Prepare fresh Fluo-4 AM loading solution: Add 25 µL of 100x concentrated detergent solvent, then add 2.5 µL of Fluo-4 AM 1000x to a 1.5 mL tube. Vortex to mix.
- Pipette 2.5 mL of culture media into the loading solution and invert to mix. Protect from light.
- Centrifuge 2 x 106 MT-4 cells at 500 x g for 3 min.
- Remove media and resuspend the pellet in 2 mL of the prepared Fluo-4 AM loading solution. Protect from light.
- Pipette resuspended cells into a 35 mm Petri dish. Incubate at 37 °C for 15-30 min, then incubate for an additional 15-30 min at room temperature.
- Transfer the cell suspension to a centrifuge tube. Centrifuge cells at 500 x g for 3 min and remove the supernatant consisting of the loading solution.
- Resuspend the cell pellet by pipetting in 1 mL of the culture medium and centrifuge at 500 x g for 3 min to wash.
- Resuspend the cell pellet by pipetting in the culture medium at a concentration of 2 x 106 cells/mL (minimum 50 µL). Protect from light.
2. Optofluidic system preparation, cell loading, and cell penning
- Prepare a chip with the wetting solution, which facilitates cell penning.
- To do this, load centrifuge tubes containing 2 mL of wetting solution and 50 mL of DI water onto the instrument with a new optofluidic chip and run the Wet Chip function. This function will automatically flood the chip with the wetting solution through the system fluidics, incubate the chip at 50, °C and flush the chip with water 3 times.
- Once water flushing is done, flush the chip with 3 cycles of 250 µL of culture media.
- Supplement the cell suspension from step 1.8 with 1:100 F-127 detergent solute before the loading to reduce the likelihood of cells sticking to the chip channels.
- Use the instrument export needle to import cells from a 1.5 mL centrifuge tube using the Load operation and Small Volume Import for a 5 µL cell package volume.
- Pen cells using an optimized optoelectronic positioning (OEP) voltage of 4.3 V and 5 µm/s cage speed. Penning occurs using OEP. Accomplish penning using the Autopen function, which will autodetect single cells, surround them with an OEP cage, and move them into a nearby pen. If cells of interest remain following auto-penning, use the Manual pen function to select target cells (with a mouse click) and a destination pen (with a subsequent mouse click).
- Once penning is complete (either when the Autopen function completes or when the desired number of penned cells has been attained), flush the chip with 3 cycles of 250 µL of culture media to clear any remaining unpenned cell from the chip.
3. Single-cell infection and functional imaging
- After penning cells, obtain fluorescent images of cells in FITC, Texas Red, and DAPI channels to measure the baseline Fluo-4, mCherry, and autofluorescence.
- Infuse HIV-1 into the microchip at a concentration of 13 ng of HIV-1 NL-CI per 2 x 106 cells by slowly pipetting the suspension into the chip through the export needle.
- Immediately after HIV-1 addition, repeatedly obtain images in the FITC and DAPI channels over a 10-min time course.
- Obtain images in the Texas Red and DAPI channels at 1-, 2-, 3-, and 4-days post-infection (DPI).
4. Analysis of single-cell functional imaging data with FIJI software
- At each time point in each imaging channel of interest, use the FIJI Point Selection tool and the Measure function to measure each cell's Fluo-4, mCherry, and autofluorescence signals. Simultaneously measure the background fluorescence by calculating the mean fluorescence intensity (MFI) of the pen and chip containing each cell.
- Control for the background fluorescence and autofluorescence by using the Measure function to find the MFI in a Region Selection and subtracting this value from the cell fluorescence value.
- Normalize the background fluorescence of each chip image: Measure the MFI of the chip in each field. Then subtract the MFI of the chip with the least background fluorescence from all other chip MFI measurements.
- Normalize the background fluorescence of each cell-containing pen: measure the MFI of each cell's pen in each field of view. Then subtract this value from the cell's raw MFI for each field.
- Control for the cell autofluorescence: Subtract the day 0 and day 4 DAPI MFI of each cell from the day 0 and day 4 mCherry MFIs.
Representative Results
Figure 1 illustrates the format of the chip and raw imaging data acquired through the fluorescence microscope. The identification and clustering of uninfected and infected cells via mCherry measurement are shown in Figure 2. These clusters are analyzed for calcium influx kinetics in Figure 3, which demonstrates early calcium influx in HIV-infected cells. Figure 4 shows a significant positive correlation between calcium influx and mCherry fluorescence 20.
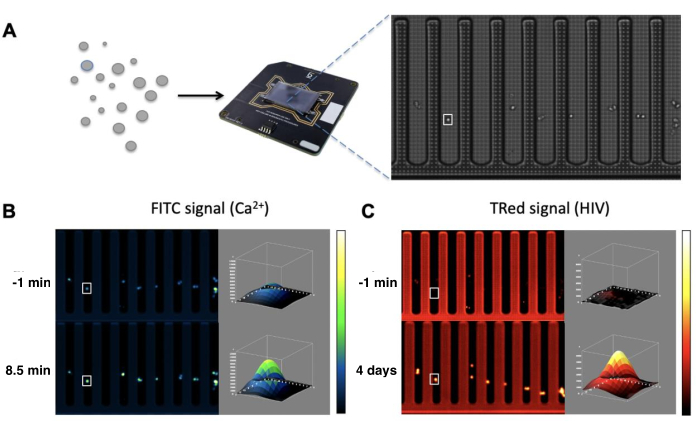
Figure 1: Chip loading and raw imaging data. Schematics of loading cells into pens on the chip using opto-electropositioning (A). Calcium influx is quantified by detecting the Fluo-4 calcium-sensitive dye in the FITC channel over an 8.5-min time course (B). HIV-1 productive infection is measured over a 4-day time course by detecting the mCherry reporter virus HIV NL-CI in the Texas Red channel (C). Please click here to view a larger version of this figure.
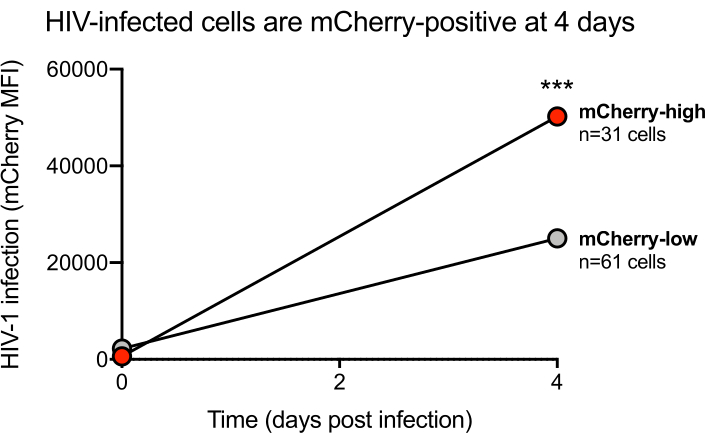
Figure 2: Representative HIV-infected cells positive for mCherry signalsat day 4 post-infection. MT-4 cells were infected with the mCherry reporter virus HIV-1 NL-CI on the optofluidic platform, and mCherry signal was measured 4 days post-infection (dpi). Cells with a change in mCherry signal >40,000 MFI (where a clear breakpoint was noted) at 4 dpi were clustered into the "mCherry-high" population; cells with a change in mCherry fluorescence <40,000 MFI were clustered into the "mCherry-low" population. Statistical software was used to plot the mean values of mCherry fluorescence at 0 and 4 dpi. Statistical significance at 4 dpi was measured using an unpaired t-test of the means (mCherry-high=50235 MFI; mCherry-low=25017 MFI; p<0.000001). *=p<0.05, **=p<0.01, ***=p<0.001 Please click here to view a larger version of this figure.
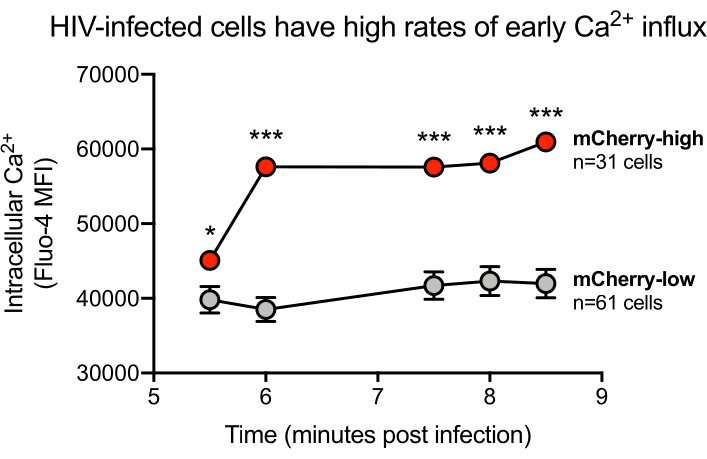
Figure 3: Representative HIV-infected cells having high rates of early calcium influx. MT-4 cells were infected with the mCherry reporter virus HIV-1 NL-CI on the optofluidic platform. Intracellular calcium was measured by Fluo-4 fluorescence repeatedly over a 9-min time course. Statistical software was used to plot the mean Fluo-4 values of the mCherry-high and mCherry-low cell clusters. Statistical significance was measured using unpaired t-tests at each time point. *=p<0.05, **=p<0.01, ***=p<0.001. Please click here to view a larger version of this figure.
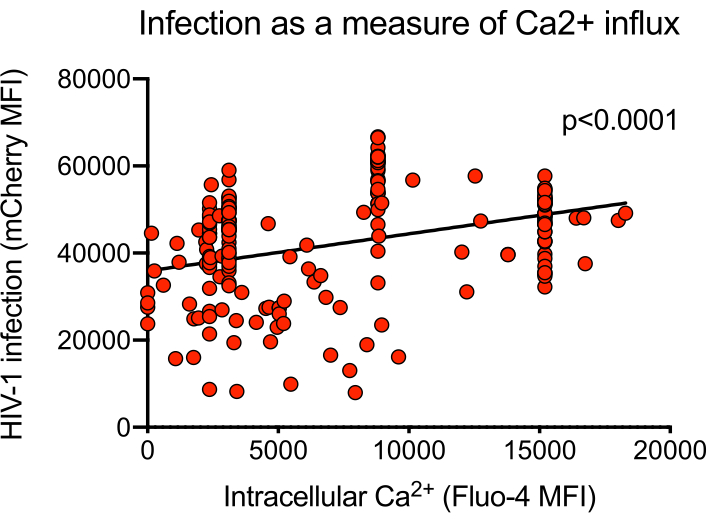
Figure 4: Representative intracellular calcium flux higher in mCherry-positive cells. MT-4 cells were infected with the mCherry reporter virus HIV-1 NL-CI. Intracellular calcium was measured by Fluo-4 fluorescence 9 min post-infection, and mCherry was measured 4 days post-infection. A simple linear regression was performed with statistical software. Please click here to view a larger version of this figure.
Discussion
The described methodology to study the relationship between calcium influx and HIV-1 productive infection in single cells can be adapted to study intracellular calcium kinetics in response to other agonists or antagonists of interest. The preparation of cells for imaging is simple, minimally time-intensive, and reagents are available in convenient kits from widely used manufacturers. Fluo-4-based calcium measurement is well-described in the literature, and HIV-1 can easily be substituted for other viruses or compounds of interest to study on this platform. Different cell types and culture mediums can be used. Alternative or additional fluorescence channels are supported, and multiple fluorophores can be imaged simultaneously or sequentially.
Additionally, the procedure can be widely adapted to study fluorophore-labeled events in longitudinal time courses. Standard high-throughput methods, such as flow cytometry, do not allow for the paired study of discrete events over time courses. Traditional single-cell imaging through fluorescence microscopy is limited by throughput, cell migration across fields of view, and intensive time requirements. By combining fluorescence imaging with high-throughput and automated single-cell culture, this method allows for the elucidation of relevant molecular kinetics, sequelae, and interactions at both short- and long-term time intervals. It is important to note that software and image analysis optimization is critical for each application.
The microscope is equipped with brightfield and Cy5, Texas Red, FITC, and DAPI fluorescence channels16. Available magnifications are 4x and 10x. This method is not currently suitable for investigations requiring more numerous fluorescence parameters or greater magnifications. Compounds of interest that will be infused into the chip for the cell exposure must exhibit diffusion to enter pens and reach cells. Access to a Beacon instrument may pose a cost barrier to investigators.
The optofluidic system has a wide scanning and stitching ability. The optical sorting on the chip has a high-efficiency variable depending on cell type, but automatic sorting efficiency is between 80-90%, and manual sorting can increase that efficiency to 100%. Each chip can accommodate 3,500 cells, and four chips can be run simultaneously, with a parallel workflow, resulting in high scalability. While the imaging workflow is reported by manual image analysis, more recent updates in the system allow for automatizing this. It is important to note that the data shown here are representative within the limitations of image threshold analysis, and optimization and automatization of image analysis are likely to result in more sensitive measures of both calcium influx and mCherry signal as indicative of HIV-1 productive infection.
The described protocol was established using MT-4 cells and HIV-1 as previously described7,21,22. When adapting the protocol to other cell lines or fluorescent targets of interest, optimization and validation are required. The kinetics of Fluo-4 efflux is cell line-dependent, so the cell type-specific dye retention should be confirmed to last at least if the intended calcium imaging time course. Validation of dye retention and agonist response on this optofluidic device can be completed by treating cells on the chip with the growth medium containing 10 µM ionomycin, which should induce potent cellular calcium influx and Fluo-4 signal in the FITC channel. Similar controls and validations should be conducted for any agonists/antagonists to be infused into the chip to ensure proper concentration and diffusion to cells. For single-cell analysis, cells should be prepared as a single-cell suspension to facilitate the sorting of individual cells into pens. Like traditional fluorescence microscopy, fluorescence exposure times and time points measured should be optimized to each respective target of interest.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We are grateful for the scientific discussions with Dr. Benjamin Chen. This work was funded by K08AI120806 (THS), R01DA052255 (THS and KB), and R21AI152833 (THS and KB).
Materials
| Beacon Optofluidic System | Berkeley Lights | ||
| Fetal Bovine Serum | Gibco | ||
| FIJI | Open-source software | PMID: 22743772, 22930834 | |
| Fluo-4 Calcium Imaging Kit | Thermo Fisher | F10489 | |
| HIV-1 NLCINL4-3 | Laboratory of Benjamin Chen | PMID: 28148796 | |
| Hyclone Pennecillin Streptomycin solution | GE Healthcare Life sciences | SV30010 | |
| MT-4 cells | NIH AIDS Reagent | ARP-120 | |
| OptoSelect 3500 chip | Berkeley Lights | ||
| Pipettor Tips | Denville Scientific | P3020-CPS | |
| Prism 9.0.0 | GraphPad | ||
| RPMI-1640 Medium | Sigma-Aldrich | R8758 | |
| Serological Pipettes | Fisher Brand | 13-678-11E | |
| Tissue Culture Hood | Various models | ||
| T75 flasks | Corning | 3073 | |
References
- Aberg, J. A. Aging, inflammation, and HIV infection. Topics in Antiviral Medicine. 20 (3), 101-105 (2012).
- Deeks, S. G., Tracy, R., Douek, D. C. Systemic effects of inflammation on health during chronic HIV infection. Immunity. 39 (4), 633-645 (2013).
- Swartz, T. H., Dubyak, G. R., Chen, B. K. Purinergic receptors: Key mediators of HIV-1 infection and inflammation. Fromtiers in Immunology. 6, 585 (2015).
- Giroud, C., Marin, M., Hammonds, J., Spearman, P., Melikyan, G. B. P2X1 receptor antagonists inhibit HIV-1 fusion by blocking virus-coreceptor interactions. Journal of Virology. 89 (18), 9368-9382 (2015).
- Hazleton, J. E., Berman, J. W., Eugenin, E. A. Purinergic receptors are required for HIV-1 infection of primary human macrophages. Journal of Immunology. 188 (9), 4488-4495 (2012).
- Marin, M., et al. High-throughput HIV-cell fusion assay for discovery of virus entry inhibitors. Assay and Drug Development Technology. 13 (3), 155-166 (2015).
- Soare, A. Y., et al. P2X Antagonists inhibit HIV-1 productive infection and inflammatory cytokines interleukin-10 (IL-10) and IL-1β in a human tonsil explant model. Journal of Virology. 93 (1), 01186 (2019).
- Sorrell, M. E., Hauser, K. F. Ligand-gated purinergic receptors regulate HIV-1 Tat and morphine related neurotoxicity in primary mouse striatal neuron-glia co-cultures. Journal of Neuroimmune Pharmacology. 9 (2), 233-244 (2014).
- Swartz, T. H., Esposito, A. M., Durham, N. D., Hartmann, B. M., Chen, B. K. P2X-selective purinergic antagonists are strong inhibitors of HIV-1 fusion during both cell-to-cell and cell-free infection. Journal of Virology. 88 (19), 11504-11515 (2014).
- Soare, A. Y., et al. P2RX7 at the host-pathogen interface of infectious diseases. Microbiology and Molecular Biology Reviews. 85 (1), (2021).
- Chen, P., Hübner, W., Spinelli, M. A., Chen, B. K. Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. Journal of Virology. 81 (22), 12582-12595 (2007).
- Dale, B. M., et al. Cell-to-cell transfer of HIV-1 via virological synapses leads to endosomal virion maturation that activates viral membrane fusion. Cell Host Microbe. 10 (6), 551-562 (2011).
- Gee, K. R., et al. Chemical and physiological characterization of fluo-4 Ca(2+)-indicator dyes. Cell Calcium. 27 (2), 97-106 (2000).
- He, M. L., Zemkova, H., Koshimizu, T. A., Tomić, M., Stojilkovic, S. S. Intracellular calcium measurements as a method in studies on activity of purinergic P2X receptor channels. American Journal of Physiology and Cell Physiology. 285 (2), 467-479 (2003).
- Li, H., Zony, C., Chen, P., Chen, B. K. Reduced potency and incomplete neutralization of broadly neutralizing antibodies against cell-to-cell transmission of HIV-1 with transmitted founder envs. Journal of Virology. 91 (9), 02425 (2017).
- Beaumont, K. G., et al. Multiparameter cell characterization using nanofluidic technology facilitates real-time phenotypic and genotypic elucidation of intratumor heterogeneity. bioRxiv. , 457010 (2018).
- Le, K., et al. Assuring clonality on the beacon digital cell line development platform. Biotechnology Journal. 15 (1), 1900247 (2020).
- Mocciaro, A., et al. Light-activated cell identification and sorting (LACIS) for selection of edited clones on a nanofluidic device. Communications in Biology. 1, 41 (2018).
- Winters, A., et al. Rapid single B cell antibody discovery using nanopens and structured light. MAbs. 11 (6), 1025-1035 (2019).
- Wu, N., Nishioka, W. K., Derecki, N. C., Maher, M. P. High-throughput-compatible assays using a genetically-encoded calcium indicator. Science Reports. 9 (1), 12692 (2019).
- Esposito, A. M., et al. A high-throughput cre-lox activated viral membrane fusion assay to identify inhibitors of HIV-1 viral membrane fusion. Journal of Visualized Experiments. (138), e58074 (2018).
- Soare, A. Y., et al. P2X1 selective antagonists block HIV-1 infection through inhibition of envelope conformation-dependent fusion. Journal of Virology. 94 (6), 01622 (2020).
