AAV Deployment of Enhancer-Based Expression Constructs In Vivo in Mouse Brain
Summary
This protocol describes a novel rAAV-based transient enhancer-reporter assay. This assay can be used to induce enhancer-driven expression in vivo in the mouse brain.
Abstract
Enhancers are binding platforms for a diverse array of transcription factors that drive specific expression patterns of tissue- and cell-type-specific genes. Multiple means of assessing non-coding DNA and various chromatin states have proven useful in predicting the presence of enhancer sequences in the genome, but validating the activity of these sequences and finding the organs and developmental stages they are active in is a labor-intensive process. Recent advances in adeno-associated virus (AAV) vectors have enabled the widespread delivery of transgenes to mouse tissues, enabling in vivo enhancer testing without necessitating a transgenic animal. This protocol shows how a reporter construct that expresses EGFP under the control of a minimal promoter, which does not drive significant expression on its own, can be used to study the activity patterns of candidate enhancer sequences in the mouse brain. An AAV-packaged reporter construct is delivered to the mouse brain and incubated for 1-4 weeks, after which the animal is sacrificed, and brain sections are observed under a microscope. EGFP appears in cells in which the tested enhancer is sufficient to initiate gene expression, pinpointing the location and developmental stage in which the enhancer is active in the brain. Standard cloning methods, low-cost AAV packaging, and expanding AAV serotypes and methods for in vivo delivery and standard imaging readout make this an accessible approach for the study of how gene expression is regulated in the brain.
Introduction
Enhancers are genomic cis-regulatory elements that serve as transcription factor binding sites and can drive the expression of a target gene in a spatiotemporally specific manner1,2. They are differentially active in different cell types, tissues, and stages of development and can be substrates of disease risk-related genomic variation3,4. Thus, the need to understand the dynamics of enhancer function is critical to progress in both translational and basic science applications within genomics. In silico predictions of enhancer activity can serve as excellent resources for generating hypotheses as to enhancer capability5,6. Such predicted enhancer activity can require additional validation and interrogation for a full understanding of the functional activity. Enhancer reporter assays have proved valuable for this purpose across a variety of systems, from cells to animals7,8,9. Towards extending these studies in a flexible and cost-effective transient in vivo context, this protocol describes the use of in vivo AAV-based methods to test putative enhancer sequences for their ability to drive the expression of an ectopic reporter gene in the postnatal mouse brain. This family of methods has utility for interrogating single candidate sequences or parallel library screening and is relevant for basic and translational research.
This method combines in a single plasmid a putative enhancer candidate DNA sequence with a reporter gene (here EGFP), under the control of a minimal promoter that alone does not drive significant expression. The plasmid is packaged into recombinant AAV (rAAV) and injected into an animal model. While the application here is to the brain, various rAAV serotypes enable infection across different tissue types so that this approach can be extended to other systems10. After a period of time, the brain can be collected and assayed for the expression of the reporter gene. Strong expression, compared with controls, indicates that the tested candidate sequence was able to "enhance" the expression of the gene (Figure 1). This simple design offers an easy and clear approach to test a sequence for enhancer activity in vivo in the brain.
In addition to testing for enhancer capability of a sequence, this method can be combined with techniques to determine cell-type enhancer activity. In sequence-based approaches to determining differential enhancer activity, sorting cells on cell-type-specific markers prior to DNA and RNA sequencing can allow researchers to determine if different cell types show differential enhancer activity, as was described in Gisselbrecht et al.11. In imaging-based approaches, co-labeling images with cell-type-specific markers allows examination of whether cells exhibiting enhancer-driven fluorescence also display cell-type markers of interest12,13,14,15,16. Enhancer reporter assays enable direct testing of risk-associated allelic variation in enhancers for effects on enhancer capability. The vast majority of risk loci identified in genome-wide association studies (GWAS) lie in non-coding regions of the genome17. Functional annotation studies of these risk loci indicate that a large portion likely act as enhancers18,19,20. MPRA deployment in vivo can allow testing of these risk-associated variants for enhancer activity in brain12,21. Finally, delivery and collection at different time points can offer insights into the developmental stages during which an enhancer is active.
Enhancer-reporter plasmid designs are diverse and can be customized to suit experimental goals. There are several options for minimal promoters that have been used in enhancer research, such as the human β-globin minimal promoter22 and the mouse Hsp68 minimal promoter23. These promoters are known to drive low levels of expression unless coupled with an enhancer element to activate them. In contrast, constitutive promoter elements drive strong expression of the transgene, useful for positive control or to test for enhancer function against a background of robust expression. Common choices for constitutive promoters include CAG, a hybrid promoter derived from the chicken β-actin promoter and the cytomegalovirus immediate-early enhancer24, or human EF1α25. Since enhancers are known to work bidirectionally26, the orientation and location of the enhancer relative to the minimal promoter are flexible (Figure 2A). Traditional enhancer-reporter assays place the enhancer upstream of the promoter and, in library deliveries, include a barcode sequence downstream of the reporter gene to associate sequencing reads with the tested enhancer27. However, enhancers can also be placed in the open reading frame of the reporter gene and serve as their own barcode sequence, as is done in STARR-seq28. The protocol described here utilizes the STARR-seq assay design, placing the candidate enhancer sequence into the 3' UTR of the reporter gene. While the STARR-seq orientation offers the benefit of more streamlined cloning, it is less well understood than the conventional approach and may induce variable RNA stability between constructs. The described methods can be easily adapted to either the STARR-seq or conventional orientation with minor alterations to the cloning process that have been described elsewhere27,29.
Different methods of AAV delivery can be employed to further customize this technique to fit experimental goals (Figure 2B). Direct intracranial injections, described further in this protocol, deliver a high concentration of virus directly to the brain30. This gives a high transduction efficiency centered at the site of injection, making this an excellent technique for experiments looking to maximize the density of transduced cells over an area of tissue. Stereotactic injection can help standardize the site of injection across animals for reproducible localized transduction. Intracranial injections are most straightforward in early postnatal animals. As an alternative technique, systemic injections can deliver transgenes using AAVs with serotypes capable of crossing the blood-brain barrier31. Tail-vein injections allow the virus to circulate throughout the body, enabling generalized delivery across many tissues10. Retro-orbital injections are another systemic injection technique that delivers the virus behind the eye into the retro-orbital sinus32. This offers a more direct route for the AAV from the venous system to the brain, resulting in a higher concentration of transduced cells in the brain than injections into more peripheral blood vessels33.
Another flexible aspect of this technique is the method of readout. Broadly, options can be described as reporter-based or sequencing-based (Figure 2C). Incorporating a fluorescent reporter such as GFP into the open reading frame of the construct results in the expression of the fluorescent protein in any transduced cells where the candidate enhancer drove expression. Labeling and imaging techniques such as immunohistochemistry enable signal amplification. Sequencing-based readout techniques involve identifying sequences from the delivered construct in RNA collected from the tissue. By quantifying the amount of viral DNA that was initially delivered, the comparison of expressed RNA versus delivered DNA can be used to determine the degree to which a tested enhancer sequence was capable of driving increased expression of the transgene, for example, in the context of a massively parallel reporter assay (MPRA). MPRAs offer a powerful expansion of these techniques to test up to thousands of candidate enhancers for activity simultaneously and have been described extensively in genomics research12,27,34,35,36. Higher throughput screening is achieved by executing cloning, packaging, delivery, and sequencing steps for candidate enhancers in batch rather than individually.
Candidate enhancer selection provides another opportunity for flexibility (Figure 2D). For example, this assay can be used to identify enhancers of a specific gene, to ascertain the function of non-coding DNA regions of interest, or to determine specific cell types or developmental stages during which an enhancer is active – all of which serve goals both in basic science and in disease research. Generally, candidate enhancer selection is driven by in silico predictions of enhancer activity. Commonly, in silico predictions include ChIP-seq for histone modifications that indicate likely enhancers, such as H3K27ac37 and chromatin accessibility mapping38. Finally, a growing area of research is the function-based screening of synthetically designed enhancer elements, enabling studies of how enhancer sequence directs function39 and the design of enhancers with specific properties40.
Protocol
This protocol has been approved by the UC Davis Institutional Animal Care and Use Committee (Protocol #22339) and the UC Davis Institutional Biosafety Committee (BUA-R1903). This protocol has been tested on C57BL/6J mice of both sexes at postnatal day 0-1.
1. Clone the enhancer candidate sequence into the AAV vector plasmid.
NOTE: The representative protocols are given, but the cloning strategy has a high degree of flexibility.
- Select or design a reporter construct. Here, the enhancer candidate is inserted into the 3' untranslated region (UTR) of the EGFP reporter gene, which is expressed under the control of the Hsp68 minimal promoter.
NOTE: Many MPRA plasmid constructs appropriate for this assay have been deposited to Addgene41 and are available for research use. - Design PCR primers to amplify a sequence of interest. Add to the 5' end of the primers the appropriate homology arm for Gibson cloning (~35 bp corresponding to sequence 5' upstream of the insertion location, top strand for the forward primer, and bottom strand for the reverse primer).
NOTE: Ensure that the base primer sequence is ~18-24 bp long and ~50%-60% GC content with a melting temperature of approximately 57-59 °C. - Perform PCR from a DNA sample using the designed primers.
- Assemble the PCR reaction mix in 0.2 mL PCR tubes as described in Table 1.
- Place the PCR reaction mix(es) on a thermal cycler and cycle according to the program mentioned in Table 1.
- Confirm the success of PCR by running 5 µL of PCR product on a 0.7%-1% agarose gel at 100 V for 30-60 min. Check for the fragment of predicted length.
- Cleanup the PCR product using a commercial enzymatic reaction cleanup column kit. The final eluate is the cloning insert.
- Perform restriction digestion to linearize the AAV vector at the unique cloning site to insert the enhancer into the vector.
- The cloning site in the 3' UTR of the enhancer reporter construct used here is bounded by unique PacI and AscI restriction sites. Perform a digest to linearize the plasmid at this location according to the following parameters (steps 1.4.2-1.4.4).
- Digest 1-10 µg of vector DNA using PacI and AscI, depending on how many cloning reactions will be needed. Prepare the reactions using the supplied buffer according to the manufacturer's instructions.
- Incubate at 37 °C for 1-2 h to digest the vector DNA. (If digesting more than 5 µg in a 50 µL reaction, longer incubation may be necessary.)
- After 1-2 h of incubation, inactivate the enzyme by incubating at 65 °C for 20 min.
- Separate the restriction digest fragments by gel electrophoresis using a 0.7%-1% agarose gel, run at 100 V for 30-60 min.
- Excise the band for the linearized vector and purify using a commercial gel extraction kit.
- Perform Gibson cloning reactions and transform
- Determine the concentration of the purified vector and the cloning insert using a spectrophotometer.
NOTE: DNA absorbs light at 260 nm, while contaminants absorb light at 230 nm and 280 nm. High ratios (>1.8) of 260/230 and 260/280 indicate highly purified DNA. - Assemble Gibson cloning reactions in 0.2 mL PCR tubes: 5 µL of 2x Gibson master mix, 0.02-0.5 pmol DNA fragments in a ratio of 3:1 insert to vector (Optimal DNA concentration for the reaction should be determined empirically) and Nuclease-free water (bring up the volume to 10 µL).
- Incubate Gibson reactions at 50 °C for 20 min in a thermal cycler.
- Determine the concentration of the purified vector and the cloning insert using a spectrophotometer.
- Transform recombination deficient (recA–) competent Escherichia coli (E. coli).
- Set a water bath to 42 °C and thaw the commercially available competent cells on ice.
NOTE: This step can be done prior to step 1.4, and the cells can thaw while preparing and incubating the Gibson reaction. - Aliquot 30-50 µL of cells into 2.0 mL microcentrifuge tubes. Add 2 µL of Gibson reaction to an aliquot and label the tube with the identity of the Gibson. Incubate the cells on ice for 30 min.
NOTE: Use 5-10 ng of undigested empty vector as a positive control and water as a negative control. - Heat-shock the cells in the 42 °C water bath for 30-45 s, immediately return the tubes to ice, and let them recover for 5 min.
- While on ice, add 400-950 µL of the commercially available recovery media.
NOTE: The recovery media used in these experiments contains 2% vegetable peptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, and 20 mM glucose. - After recovery on ice, fully recover the cells by incubating at 37 °C with gentle agitation of 250 RPM for 30-60 min.
- Pellet the cells by centrifugation at 5,000 x g for 5 min and remove 400 µL of the supernatant, leaving ~50 µL of bacteria to plate.
- Plate on selective media and incubate at 37 °C overnight.
NOTE: The selective media used in these experiments contains 1.0% tryptone, 0.5% yeast extract, 1.0% sodium chloride, 1-2% agar, 96-97% water, and carbenicillin at a concentration of 100 µg/mL. Always use recombination deficient strains of bacteria for cloning viral plasmids because the viral inverted terminal repeats (ITRs) have matching sequences and can undergo homologous recombination. However, recA– strains of bacteria still undergo low levels of recombination. The reporter plasmid used here is a self-complementary AAV, where the sequence of one of the ITRs has been altered and no longer perfectly matches the other ITR. Growing the bacteria at 30 °C is also recommended for further slowing the rate of homologous recombination.
- Set a water bath to 42 °C and thaw the commercially available competent cells on ice.
- Verify clones and recover plasmids.
- Prepare a PCR master mix using forward and reverse primers (Table of Materials) that anneal to the vector, flanking the insert location. Aliquot 10 µL volumes into 0.2 mL PCR strip tubes.
- In 14 mL round-bottom tubes, prepare 5 mL aliquots of antibiotic selective LB broth, one for each colony being tested via PCR and negative control.
- Use a sterile toothpick or pipette tip to pick an individual colony and roughly scrape the colony down into the bottom of one PCR tube.
- When the colony has dissociated into the PCR master mix, drop the toothpick or tip into one of the LB aliquots and label the 14 mL tube with the Gibson reaction and the PCR tube number.
- Place the colony PCR reactions on a thermal cycler and cycle with the program described in Table 2.
- Incubate the 5 mL liquid cultures inoculated with toothpicks or pipette tips in a shaking incubator set to 37 °C and 180 RPM.
- Run the colony PCR reactions on a 0.7%-1% agarose gel at 100 V for 30-60 min and visualize the bands in a gel doc imaging system.
- Discard the 5 mL cultures that do not display the predicted PCR band.
- For each successfully integrated clone, allow the 5 mL cultures to grow overnight and then perform a plasmid mini prep from 4.5 mL using a commercial kit. Reserve 0.5 mL of the liquid culture and save at -80 °C as a glycerol stock.
- Confirm that the transgenes contained within the plasmids are free of undesirable mutations using Sanger sequencing42.
- After the reporter construct has been confirmed to be successfully cloned, use the saved glycerol stock to inoculate a 5 mL starter culture and grow for 8-16 h in a shaking incubator at 30 °C and 180 RPM.
- Once the broth is cloudy, dilute the starter culture 1,000x in 250-300 mL of fresh selective LB broth and incubate at least overnight in a shaking incubator at 30 °C and 180 RPM. Then purify the plasmid using a commercial plasmid maxi kit.
NOTE: Bacteria will grow more slowly at 30 °C compared to 37 °C, but this is the best temperature for slowing the rate of spontaneous recombination when growing large cultures. - Perform an XmaI digest to test for recombination of the ITRs using steps 1.4.2-1.4.5, substituting XmaI instead of PacI and AscI. Visualize the bands on a gel doc imaging system.
NOTE: An rAAV plasmid with both ITRs present will yield two bands on a gel following this digest: one the length of the rAAV genome and the other the length of the rest of the plasmid backbone. If there is only one band, then the ITRs have undergone homologous recombination, and the rAAV plasmid will not be capable of packaging viral particles. The rAAV plasmid backbone described here has been designed so that XmaI sites only occur in the ITRs. If the enhancer candidate insert also contains any XmaI sites, update the predicted band results and analysis accordingly.
2. Obtain packaged rAAV.
- Use packaged rAAV (professionally or in-house) for the experiments in this study.
NOTE: There are many options for packaging an rAAV. An rAAV can be professionally packaged at a company or a university core facility. The rAAV can also be packaged in-house using different techniques that range in complexity and need for specialized equipment43,44. The primary concern is obtaining a vector that is high quality and that the infectious titer has been determined to a high degree of certainty. Both professionally packaged and in-house packaged rAAVs were used for different experiments described in this study.
3. Intracranially inject rAAV-packaged plasmid into neonatal mice.
- Prepare rAAV mixture for delivery.
- If the intent is to compare the expression patterns between different enhancer reporter viruses, equalize the titers of all viruses to be compared by diluting all to match the least concentrated virus.
- Mix together a 2:1 or 3:1 ratio of enhancer reporter virus and constitutively expressed control reporter virus and add a small amount (0.06% final concentration) of Fast Green dye.
NOTE: Fast Green is a dye that is used widely in injections of experimental animals, including with AAV, to visualize the injection site during and immediately after the procedure45,46,47,48. While this is an important quality assurance step, it is possible that the addition of dye could interfere with transduction. Reconsidering the use of injection dye may be important for protocol optimization in certain cases. The constitutively driven control virus is absolutely critical for comparing independent injections. Virus injections will vary in location and extent of transduction from animal to animal. The constitutive control will enable excluding failed injections from analysis and provide a standard for normalization of variation in virus delivery. - Break the tip of a sterile pulled glass pipette made from a μL-graduated capillary tube by gently piercing it through a delicate wipe sterilized by soaking it in 70% ethanol and allowed to dry completely. Then insert the pulled glass pipette into the aspirator assembly consisting of a device for applying pressure connected to 15-inch long rubber tubing ending with a rubber gasket for holding a micro-capillary pipette.
NOTE: The "device for applying pressure" may be a syringe or a mouthpiece. If a syringe is to be used, a second person will be required to apply pressure through the syringe while the experimenter manipulates the pipette in one hand and the animal in the other. If using a mouthpiece, enabling the experimenter fine control over both the positions of the animal and pipette and the pressure applied to the syringe, extra care to avoid virus contamination is required. - Apply negative pressure through the aspirator assembly to draw a small volume (~0.2-0.5 µL) of mineral oil into the glass pipette.
NOTE: This step is very important to provide a barrier to prevent aerosolized viral particles from contaminating the aspirator assembly. - Set the prepared aspirator assembly aside and keep the virus on ice until ready to inject.
- Cryo-anesthetize the neonatal mice in a dry plastic dish partially submerged in ice until the cessation of the pedal withdrawal reflex (~5 min). Ensure that the mice are not in direct contact with the ice.
- Use negative pressure through the aspirator assembly to draw the virus mixture into the pulled glass pipette until the meniscus passes the 2 µL tick mark.
- Remove the mouse from the cold chamber and position it on the benchtop. Locate the bilateral injection sites midway between lambda and bregma and midway between the sagittal suture and each eye. Sanitize both areas with an alcohol wipe.
- Use the pulled glass pipette to pierce the skull of the neonatal mice and gently apply positive pressure through the aspirator assembly as the needle enters the brain. Watch the meniscus of the virus mixture. Resistance to the pressure applied will decrease when the tip of the pipette reaches the lateral ventricle. Dispense 1 µL of the virus, withdraw the pipette, and repeat for the other lateral ventricle.
- Place the mouse on a heating pad or a warming chamber to recover. Once awake and active, return the animal to its home cage.
4. Collect tissue and perform immunohistochemistry.
- After sufficient time following virus transduction, anesthetize the mice and sacrifice by transcardial perfusion.
NOTE: Many experiments, including the representative results shown here, allow a period of 4 weeks or longer for the virus to transduce. However, self-complementary AAVs can show strong expression as early as 7 days12. The desired incubation period may need to be optimized depending on specific experimental techniques and goals.- Prepare a peristaltic pump with intravenous infusion tubing and a 25 G needle.
- Prepare 1x PBS and 4% paraformaldehyde (PFA) and place them on ice.
- Place the intake end of the tubing in the ice-cold 1x PBS. Set the flow rate (2.5 mL/min for P7 mice, 3.5 mL/min for P28 mice) and run the pump until the buffer is drawn all the way through the tubing and is cleared of bubbles. Set the needle aside until ready for perfusion.
- Inside a fume hood, pipette a small volume of isoflurane onto a paper towel in the bottom of a drop jar chamber. Place the previously injected mouse onto a grate above the paper towel, ensuring it does not come into direct contact with the isoflurane. Seal the chamber to anesthetize the mouse. Wait for ~5 min and then open the chamber and check for a pedal withdrawal reflex. Proceed once the animal is unconscious.
NOTE: Throughout the perfusion, the mouse should be maintained on isoflurane with a nose cone to prevent regaining consciousness. - Use surgical scissors to open the abdomen and chest of the mouse, remove the rib cage and expose the heart.
- Use a pair of iris micro scissors to make a small incision in the mouse's right atrium, insert the IV needle into the left ventricle, and activate the peristaltic pump.
- Perfuse the animal with ice-cold 1x PBS until the blood flushing out of the incision in the right atrium runs clear. Clearing the tissue of blood is very important because red blood cells have autofluorescence, and blood vessels may impair the ability to image reporter-expressing cells.
- Pause the peristaltic pump and move the intake tubing from the PBS to the 4% PFA. Reactivate the pump and perfuse 1 mL/g of body weight, ~25 mL for an adult mouse.
NOTE: Fixation tremors should be observable, and the body of the mouse should stiffen.
- Dissect and post-fix the brain.
- Remove the mouse's head with surgical scissors.
- Using small surgical scissors, start at the base of the skull and make an incision along the midline of the head and pull back the skin and underlying tissue to expose the skull.
- Using iris scissors, carefully cut the back of the skull, starting at the foramen magnum and continuing up toward and along the sagittal suture until the olfactory bulbs are passed.
- Insert the scissors into the skull over the olfactory bulbs and make two horizontal cuts in the skull. Make a similar pair of horizontal cuts in the occipital bone. The horizontal and midline cuts create a pair of door-like flaps in the top of the skull.
- Peel back the flaps of the skull to fully expose the brain. Use a pair of tweezers to sever the olfactory bulbs from the skull and to cut the cranial nerves, freeing the brain from the skull.
- Drop the brain into a 15 mL conical tube with 3-5 mL of 4% PFA in PBS. Post-fix 6 h to overnight. Do not over-fix the tissue.
- Prepare the brains for cryosectioning.
- Transfer the fixed brain to a new 15 mL conical tube with 12-14 mL of 30% sucrose in PBS for dehydration. Incubate at 4 °C until the brain sinks to the bottom of the tube (2-3 days).
- Submerge the fixed and dehydrated brain in optimum cutting temperature (OCT) compound in a 15 mm x 15 mm x 5 mm cryomold. Add OCT until the brain is fully covered.
- Place cryomold on a block of dry ice and freeze the sample in a solid block of OCT (~ 5 min).
NOTE: OCT blocks can be stored at -80 °C for months to years. - Label the cryomold with the sample name and the orientation of the brain.
- Obtain coronal sections using a cryostat microtome.
- Mark the orientation of the brain on the OCT block in pencil and remove the block from the cryomold inside the cryostat.
- Position the OCT block so that the brain is oriented with the olfactory bulbs facing the blade of the cryostat and adhere the block to the object holder of the cryostat with some fresh OCT.
- Once frozen in place, begin cutting 50 µm sections through the block following steps 4.4.4-4.4.6.
- Do not use the anti-roll plate. Sections will curl into tight rolls as they are cut and will collect either on top of the block or fall below into a collection tray.
- Watch the shape of the block in the first few cuts that are made. Make vertical cuts through the block, producing an even face when beginning to cut. Adjust the angle using the sample adjustment arms if needed.
- When the olfactory bulbs are visible, pay close attention to the shape of the sections. If one appears larger than the other, the cuts are at an angle relative to the brain, and it is necessary to straighten the orientation of the brain against the blade using the sample adjustment arms.
NOTE: If cutting straight, the cortex should begin to appear above each olfactory bulb at the same time. - Once the olfactory bulb has been sectioned and rostral brain tissue is visible, reduce the cutting thickness to 30-35 µm and begin to collect the floating sections. Follow steps 4.4.8-4.4.13 for sectioning the brain.
- Brush all previous sections off of the block and object holder so that they fall into the collecting tray. Brush them off to one side of the tray and keep them separate. If the pile is too big, then empty the tray.
- Dispense 1 mL of PBS into each well of a 24 well plate. Label 1 row for each brain.
- Cut 6 coronal sections. Retrieve the sections using cold tweezers and arrange them on the cryostat stage. Repeat this step 2 or 3 times, keeping the groups of 6 sections separate.
- Wet a size 000 paintbrush with PBS and dab away excess liquid on a lint-free tissue or paper towel. Use the paintbrush to gently pick up each section, one at a time, and place it in the appropriate wells of the 24 well plate.
- Place one of the 6 sections from the group of slices into each of the 6 wells in a row of the 24 well plate. This ensures that each well contains representative sections that span the sectioned area of the brain.
- Continue sectioning in groups of 6 until the caudal end of the cerebral cortex has been sectioned.
- For storage, seal the 24 well plate with parafilm and store at 4 °C.
NOTE: Sections are stable for 1-2 months at 4 °C. A PBS solution with 0.1% Sodium azide may be used to inhibit bacterial growth and prolong the shelf life of the sections. However, if the sections are to be stored for longer than 2 months, they should be placed in cryoprotectant solution and frozen at -20 °C for best results.
- Perform immunohistochemistry for reporter gene signal amplification.
NOTE: All steps except antigen retrieval should be performed using gentle-to-moderate agitation (~100 RPM) on an orbital shaker.To preserve native fluorescence of the control reporter (mRuby3), all steps should be protected from light as much as possible.- Prepare necessary buffers as described in Table 3.
- Prepare a new 24 well plate with one row for each brain being stained. In the first column of wells, add 1 mL of PBS to each well.
- Using a size 000 paintbrush, gently transfer 1 well of sections from the original collection well to the fresh PBS in the new 24 well plate for a quick rinse to remove residual OCT compound.
- Add 1 mL of ARB to the next well in the 24 well plate and transfer sections to this well. Incubate in an oven at 60 °C for 1 h.
- Allow the plate to cool at room temperature (RT) for 20 min.
- Add 1 mL of PB to the next well and transfer the sections to this well. Incubate at RT for 20 min.
- Add 500 µL of BB to the next well and transfer the sections to this well. Incubate at RT for 1 h.
- Add 2 mL of WB to the BB and then pipette out ~2 mL of solution and discard, leaving a diluted BB solution in the well. Repeat until the sections are clearly visible.
- Dilute the primary antibody (Chicken IgY anti-GFP, 1:1000) in BB. Add 350-500 µL of primary antibody solution to the next well and transfer the sections to this well. Seal the plate with parafilm and incubate at 4 °C overnight.
- Dilute BB with WB as done previously until the sections are clearly visible.
- Add 1 mL of WB to the next well and transfer the sections to this well. Incubate at RT for 20 min.
- Remove ~800-900 µL of buffer and discard. Add 1 mL fresh WB and incubate 20 min. Replace 1 mL of WB 4 more times for a total of 5 washes, 20 min each.
- Dilute the secondary antibody (Donkey anti-chicken Alexa Fluor 488 conjugated, 1:500) in BB. Add 350-500 µL of the secondary antibody solution to a new well. Incubate at RT for 45 min to 1 h.
NOTE: This is the seventh well, so if staining sections from more than 2 brains, a new plate will be needed at this point. - Dilute BB with WB as previously and transfer the sections to a new well filled with 1 mL of WB. Wash the sections as done previously for 20 min 3 times.
- Prepare a working solution of DAPI in PBS (final concentration 0.04-0.8 µg/mL). Protect solution from light.
- Add 1 mL DAPI solution to the next well and transfer the sections to this well. Incubate at RT for ~5 min.
- Transfer the sections back to WB and gently mount on microscope slides using a size 000 paintbrush. Allow the slides to air dry.
- Apply coverslips with an appropriate mounting media. Allow slides to cure at RT in the dark overnight.
5. Image and analyze brain tissue sections for enhancer activity.
- Use a low magnification (5x) objective lens to record fluorescence images that span the brain section.
- Locate the injection site and evaluate the extent of the virus transduction based on the density and intensity of red fluorescent cells. Take care to compare animals that were transduced similarly.
- Record detailed fluorescent images with a higher magnification objective lens (≥25x) if desired.
NOTE: Always be sure to use the same settings for acquisition parameters such as excitation light intensity, filters, and exposure time between samples that are to be compared. - Process the images using image analysis software.
NOTE: Processing may be done in any image analysis software package. The following steps are suggestions for determining whether a sequence acts as an enhancer in the reporter construct context (a yes or no question) using the open-source FIJI distribution of ImageJ. More in-depth photometry may be appropriate when comparing degrees of activity between enhancer variants. Images from different samples should always be processed consistently in order to be compared.- Apply filters to reduce noise. In Fiji select the Despeckle option under the Process > Noise menu.
NOTE: This is a 3 x 3 median filter. - Subtract background fluorescence following steps 5.4.3-5.4.6.
- Separate the channels of a multichannel image. In Fiji, select the option Split Channels under the Image > Color menu.
- Use the rectangle or circle selection tool to draw a small shape in an area of the image that does not have any fluorescent cells and measure the average pixel intensity of the background using Analyse > Measure. Repeat to sample multiple areas.
NOTE: Make sure Mean Gray Value is selected in the Analyze > Set Measurements menu. - Average the mean gray value from 5-8 areas of background and drop the decimal point. Subtract this value from each pixel in the image using Process > Math > Subtract.
NOTE: Rounding to the nearest whole number could over-estimate the background to subtract. It is more conservative to simply drop the decimal point. For example, 6.4 would be rounded down to 6, but 6.5 should also be rounded down to 6 to avoid the risk of subtracting 0.5 units of real signal from each pixel. - If desired, merge the channels back into a multichannel image. In Fiji, use Image > Color > Merge Channels.
- Stitch any overlapping images together. In Fiji, use Plugins > Stitching > Grid/Collection stitching.
- Adjust brightness and contrast. In Fiji, use Image > Adjust > Brightness/Contrast.
NOTE: Always use the same minimum and maximum values for each image to be compared. - Count the number of green fluorescent cells following steps 5.4.10-5.4.12. These are the cells in which the candidate enhancer sequence was able to drive reporter expression.
- Start by hiding the green channel and use the freeform selection tool to draw a shape around the region of the brain that expresses red fluorescence. Using the Measure function in Fiji, measure the area, integrated density, and mean gray value of the red channel in this zone.
- Using the multi-point selection tool, count the number of green cells in this zone.
- Normalize the number of green cells by the integrated density of the red channel. The local red channel brightness is a proxy measure for the magnitude of virus exposure, enabling the comparison of enhancer activity between different transduced animals.
- Apply filters to reduce noise. In Fiji select the Despeckle option under the Process > Noise menu.
Representative Results
Using these methods, a 915 bp sequence in the psychiatric risk-associated third intron of the gene CACNA1C19,49,50 was tested for enhancer activity in the postnatal mouse brain. This sequence was discovered in an MPRA of 345 candidate enhancer sequences centered on psychiatric and neurological risk SNPs12 and characterization experiments are described here as a general example. C57BL/6 mice were injected at P0 with an AAV9 construct packaged as a self-complementary vector51 containing EGFP under the control of the Hsp68 minimal promoter and a single candidate enhancer sequence (Figure 3A). Additional mice were injected with constructs that contained a negative control sequence that was predicted to have no regulatory activity (Figure 3B). Viral titers for these constructs were normalized to 6.85 x 1010 vg/mL. All injected mice were co-injected with an AAV9-packaged construct containing mRuby3 under the control of the constitutive CAG promoter to visualize the area that was successfully transduced and control for potential variability in the site and spread of infection. Brains were collected at postnatal day (P)28 for immunohistochemistry and imaging. These experiments confirmed that this candidate enhancer region in CACNA1C drove expression of EGFP in P28 mouse brain (Figure 3A), while a negative control sequence did not (Figure 3B). These results demonstrate the application of enhancer-reporter assays in the mouse brain, enabling in vivo study of enhancers during conditions that cannot be recapitulated using traditional in vitro models.
These methods can also be used to test libraries of candidate enhancer sequences for activity using AAV-based delivery methods, such as in MPRAs. Representative images of library-based reporter expression at different titers demonstrate the flexibility of this assay and the effect of viral titer on transduction efficiency. High-titer (Figure 4A) versus low-titer (Figure 4B) viral preparations result in differences in the amount of transduced cells, as evidenced by both positive control mRuby3 expression driven by a CAG promoter and EGFP expression produced from the candidate enhancer libraries in postnatal mouse brain. There are situations where either high titer and broad transduction or low titer and sparse transduction may be preferred.
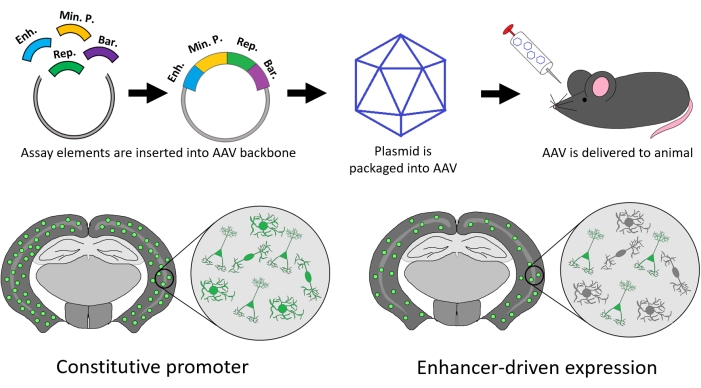
Figure 1: Overview of the protocol. Key elements of this assay include a candidate enhancer element, a minimal promoter, a reporter gene, and, depending on assay design, a barcode sequence for unique tagging. These elements are cloned into an AAV plasmid backbone and packaged into AAV. The virus is delivered to the animal and left to incubate for a number of days or weeks. Finally, the tissue of interest is collected and enhancer activity is ascertained via imaging or sequencing. Enhancer-driven transgene expression can produce expression with cell-type, regional, and temporal specificity, in contrast to constitutive drivers. Please click here to view a larger version of this figure.
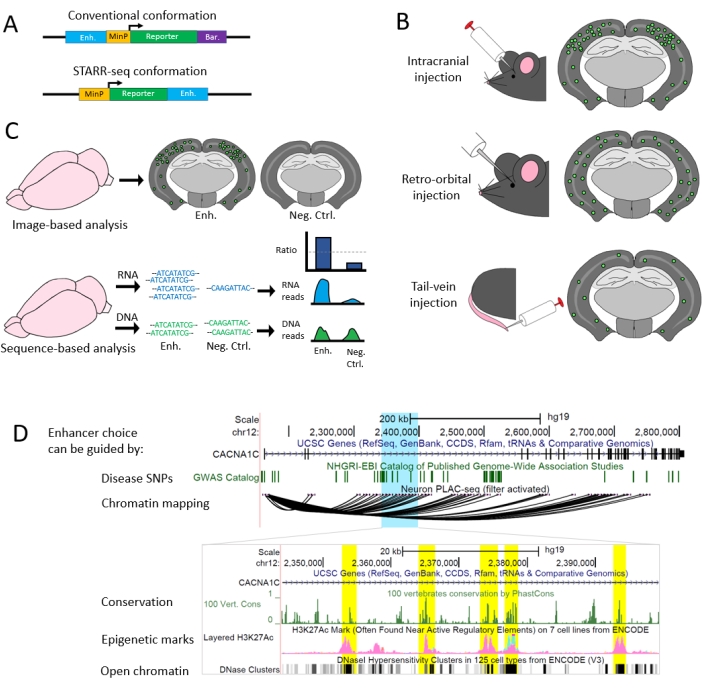
Figure 2: Flexibility in assay design. (A) Plasmid orientation can place the enhancer upstream of the minimal promoter or downstream of the reporter gene, as in STARR-seq28. For sequence-based readouts, a barcode is included downstream of the reporter gene, or the enhancer can serve as its own barcode in the second orientation. (B) Common viral delivery techniques to the brain include intracranial injections, retro-orbital injections, or tail-vein injections, which may result in different densities of transduced cells in different tissues. (C) Readouts can be sequencing-based or imaging-based. In sequencing-based readouts, enhancer activity is defined by a relative increase of RNA sequence versus input DNA sequence (controlling for AAV delivery). In imaging readouts, expression of the reporter gene is increased where the enhancer is active. (D) Potential intronic enhancers in the psychiatric risk-associated gene CACNA1C are highlighted. In the top image, a broad region is selected that shows strong associations to GWAS-tested traits and interactions with the CACNA1C promoter via PLAC-seq20. In the inset, smaller candidate regions are identified that are evolutionarily conserved at the sequence level, are in areas of open chromatin, and show epigenetic marks indicative of enhancers. Please click here to view a larger version of this figure.
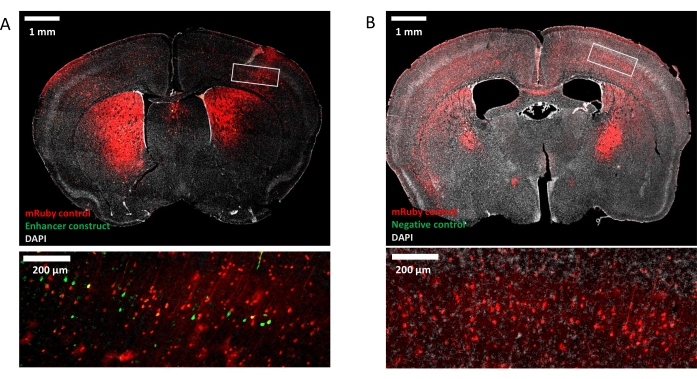
Figure 3: Enhancer in CACNA1C drives EGFP expression in P28 mouse brain. (A) A mouse injected intracranially at P0 with an enhancer-reporter construct (scAAV9-Hsp68-EGFP-CACNA1C_3) and constitutively expressed injection control (AAV9-CAG-mRuby3) shows enhancer-driven EGFP expression in the middle-lower layers of the cortex at P28. (B) A mouse injected at P0 with a negative control construct (scAAV9-Hsp68-EGFP-NEG_4) and injection control does not show expression of EGFP at P28. Whole-brain images were taken at 5x magnification and insets show zoomed-in view of the boxed region. Please click here to view a larger version of this figure.
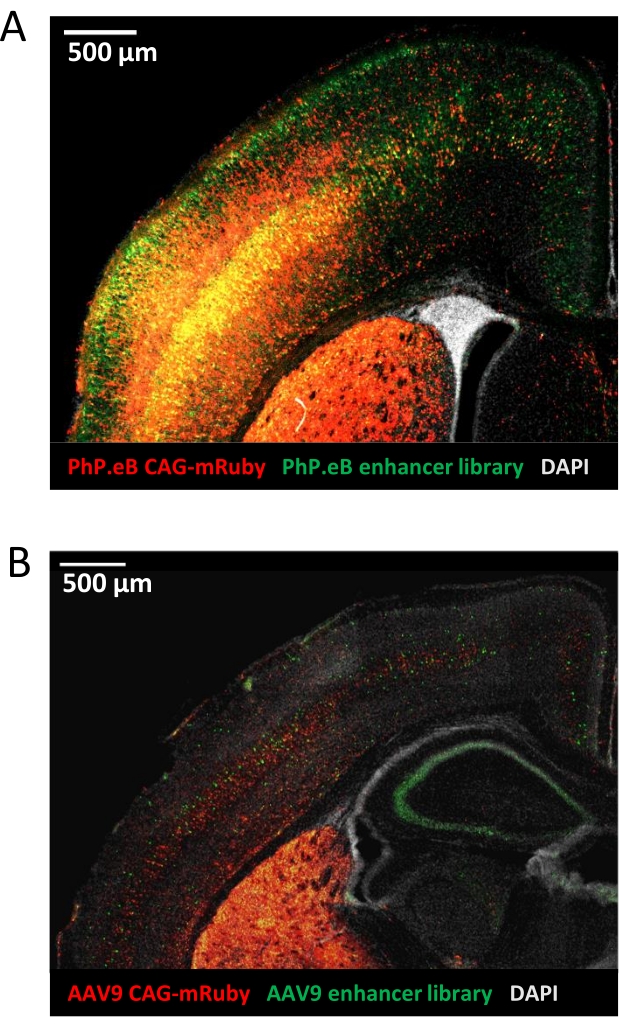
Figure 4: AAV-delivered enhancer-reporter libraries of different titers show activity in mouse brain. (A) Commercially packaged, high-titer AAV-PHP.eB library of candidate enhancers drives broad EGFP expression in P7 mouse brain. (B) Lower-titer AAV9 library of candidate enhancers precipitated from conditioned media of the packaging cells drives sparse EGFP expression in P7 mouse brain. Images were taken at 5x magnification. Please click here to view a larger version of this figure.
| PCR Reaction Mix | |
| 5x reaction buffer | 10 µL |
| dNTPs | final concentration of 200 µM each |
| Cloning primers | final concentration of 0.2 µM each |
| Template DNA | 50 ng |
| High fidelity polymerase | final concentration of 0.02 U/µL |
| Nuclease-free water | to reach 50 µL final reaction volume |
| Thermocycling conditions | |||
| 30 cycles of the following: | Notes | ||
| Step | Temperature | Time | |
| Denature | 98 °C | 10 s | |
| Anneal | 60 °C | 20 s | Optimal temperature may vary based on primers. |
| Extend | 72 °C | 30 s | Optimal temperature and duration may vary based on polymerase. |
| Final Extension | 72 °C | 2 min | Optimal temperature and duration may vary based on polymerase. |
| Hold | 4 °C | ||
Table 1: PCR reaction mix and thermocycling conditions.
| Thermocycling conditions | |||
| 30 cycles of the following: | Notes | ||
| Step | Temperature | Time | |
| Cell Lysis | 98 °C | 5 min | |
| Denature | 98 °C | 10 s | |
| Anneal | 55 °C | 15 s | Optimal temperature may vary depending on primers. |
| Extend | 72 °C | 30 s | Optimal temperature and duration may vary depending on polymerase. |
| Hold | 4 °C | ||
Table 2: Thermocycling conditions for colony PCR.
| Buffers for immunohistochemistry | |
| Buffer | Composition |
| 1x PBS | 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 |
| 1x Citrate Antigen Retrieval Buffer (ARB ), pH 6.0 | Proprietary, see table of materials |
| Permeabilization Buffer (PB) | 1x PBS + 0.5% Triton X100 |
| Wash Buffer (WB) | 1x PBS + 0.1% Triton X100 |
| Blocking Buffer (BB) | WB + 5% milk |
Table 3: Buffers for immunohistochemistry.
Discussion
This protocol describes an rAAV-based method for the deployment of enhancer-driven transgenes in the postnatal mouse brain. In this generalized protocol, a candidate enhancer, a minimal promoter, a reporter gene, and an optional barcode sequence are cloned into an AAV plasmid backbone. These experiments can be done with a single candidate enhancer sequence or with many sequences in parallel. The plasmid is packaged into an rAAV and delivered to the postnatal mouse brain. After a period of time to allow for virus transduction, the brain is collected and imaged for reporter gene expression. A constitutively expressed reporter virus (CAG-mRuby3) is included as an injection control. Using this assay, enhancer elements are shown to drive transcription of EGFP in the mouse brain, demonstrating enhancer activity in vivo.
The primary targets for troubleshooting and optimization of this assay include viral titer, injection technique, and transduction time frame. Different viral titers can have a strong effect on the density of transduced cells. While a higher transduction efficiency is likely preferable for most research, the optimal level of transduction is highly dependent on experimental goals. Intracranial injections offer a high density of transduced cells, but are likely to be more focal at the site of injection, though the spread of infection may depend on viral serotype and age of the animal52. Retro-orbital injections may show more even distribution of transduced cells throughout the brain but are less likely to give any area of high infection. Tail-vein injections can more effectively transduce other tissues in addition to the brain but will likely give still lower densities of transduced cells in the brain. Similarly, a variety of time-frames for virus transduction in the brain can serve different purposes. Robust scAAV expression can occur in the brain as early as seven days after intracranial injection, though periods of 28 days or longer are more typically used and yield strong expression. As enhancers can be temporally specific in their activity, it is important to consider the predicted activity of the tested enhancers when determining study design.
There are some limitations associated with these methods that might make other options a more suitable choice, depending on experimental goals. Since AAVs exhibit low integration into the genome, tissues are most effectively transduced when they are mature and there is a high density of postmitotic cells, as a large amount of continued cell divisions will lower the density of transgene-expressing cells. For viral delivery of a transgene to a less mature model, such as tissues that are still growing, lentiviruses may be a more appropriate choice, as they integrate into the genome and will be inherited by all daughter cells of a transduced proliferating cell. Additionally, while the reporter assay described here is a widely used technique for the functional study of cis-regulatory activity, it cannot provide a complete picture of how the regulatory element acts within its native context. For example, this assay does not provide information on what genes may be targeted by the regulatory element in the genome. However, if the enhancer's target genes are known, this information can be leveraged to determine if the cell types showing expression in this assay are the same as those known to express the enhancer's targeted genes, which would lend high biological validity to the interpretation of results. The assay also may not completely recapitulate the effects that the surrounding genomic sequence or normal biological and behavioral activity of the animal may have on the activity of the tested regulatory element. Finally, this protocol describes the STARR-seq reporter assay design, in which the candidate enhancer element is cloned downstream of the reporter gene in the ORF, in contrast to upstream of the promoter, as in the conventional reporter assay cloning orientation. Including long variable sequences in the ORF of the reporter gene may cause reduced RNA stability due to factors such as RNA binding protein motifs, alternate splice sites, or variable GC content 36,53, and statistical methods have been developed to account for sequence-based biases when analyzing STARR-seq data54,55. Despite these considerations, STARR-seq MPRAs have been widely used in recent years to successfully identify cis-regulatory elements56,57,58,59. The methods described here can be easily adapted for use with the conventional MPRA orientation.
The cell-type specificity of enhancers makes this assay a powerful tool in driving the cell-type-specific expression of genes of interest in vivo. Current techniques for cell-type-specific expression of a desired gene can be limited by the availability of conditional expression constructs or require the generation of new transgenic mouse lines. Using an rAAV-based enhancer-driven system, a gene of interest can be expressed in a spatiotemporally specific manner by using validated cell-type-specific enhancers or promoters to drive expression13,14,15. Recent developments in this technology have shown that combining cell-type-specific enhancers with rAAV serotypes with tropisms for cells or tissues of interest can show similar specificity of transgene expression as in transgenic animal models60. This offers a cheap, fast, and potentially high-throughput method for sequence screening and specific induction of transgene expression in vivo in animal models.
This technology also has therapeutic potential. Clinical studies have found that rAAV-based delivery of a transgene in vivo can induce the expression of healthy copies of a gene in cases where only a defective copy is present, or the gene is not being transcribed at a sufficient rate61,62,63. The selection of a driving enhancer element has the potential to enable generalized delivery but expression induction in a cell-type-specific manner. Finally, delivering the transgene via rAAV also confers significant benefits; AAVs have lower immunogenicity than other viruses often used in transgene delivery64, making this a potentially safe viral vector to be employed for clinical use.
Specific deployment of in vivo rAAV-based enhancer-reporter assays is customizable and can be modified to suit a range of experimental goals. Different tissues and cells can be targeted by selecting AAV serotypes or enhancers that have cell type or spatiotemporal specificity. Anywhere from one to thousands of enhancer-reporter constructs can be delivered in vivo, and activity can be assayed using a number of imaging- and sequencing-based readouts, as have been shown in an emerging set of studies using this approach15,16,65,66,67,68. The range of options for construct design and delivery enables this technique to be modified for a variety of uses in both translational and basic science, making this a powerful new tool for genomics and neuroscience research.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Sequencing was performed at the UC Davis DNA Technologies Core. We thank the lab of Lin Tian at UC Davis for training on rAAV packaging and generously gifting us AAV helper and rep/cap plasmids. This work was supported by NIH/NIGMS R35GM119831.
Materials
| 10x Citrate Buffer | Sigma-Aldrich | C9999-1000ML | |
| 5'-gatcactctcggcatggac-3' | Integrated DNA Technologies | N/A: Custom designed | Forward primer for verifying clones after transformation. These primers are specific to the vector used and were designed for the specific vector used in our experiments. |
| 5'-gatggctggcaactagaagg-3' | Integrated DNA Technologies | N/A: Custom designed | Reverse primer for verifying clones after transformation. These primers are specific to the vector used and were designed for the specific vector used in our experiments. |
| Agarose | VWR | VWRVN605-500G | |
| Aspirator tube assemblies | Sigma-Aldrich | A5177-5EA | for mouth-driven delivery of rAAV |
| Bacteriological petri dishes | Thermo Fisher Scientific | 08-757-100D | |
| Carbenicillin | Sigma-Aldrich | C1389-5G | |
| Chicken IgY anti-GFP | Thermo Fisher Scientific | A10262 | |
| Confocal microscope | Zeiss | LSM900 | The images were taken on the LSM800 model, but Zeiss launched the LSM900 model in recent years to replace LSM800. |
| Conical centrifuge tubes 15 mL | Thermo Fisher Scientific | 12-565-269 | |
| Cryomolds | Thermo Fisher Scientific | NC9806558 | These molds are suitable for P28 mouse brain. Other sizes may be more suitable for larger or smaller tissues. |
| DAPI | Sigma-Aldrich | D9542-10MG | |
| Dissecting scissors, 4.5" | VWR | 82027-578 | |
| Donkey anti-chicken AlexaFlour-488 | Jackson ImmunoResearch | 703-545-155 | |
| Dulbecco's PBS 1x | Thermo Fisher Scientific | MT21031CV | |
| Eppendorf Microcentrifuge tubes 2.0 mL | Thermo Fisher Scientific | 22431048 | |
| Falcon round-bottom tubes 14 mL | Thermo Fisher Scientific | 352059 | |
| Fast Green dye | Grainger | F0099-1G | |
| Fine detail paint brush set | Artbrush Tower | B014GWCLFO | |
| Gibson Assembly Master Mix | NEB | E2611S | |
| Glass capillary tubes | Drummond Scientific Company | 5-000-2005 | |
| HiSpeed Plasmid Maxi Kit | QIAGEN | 12663 | Commercial plasmid maxi prep kit |
| HyClone HyPure Molecular Biology Grade Water | VWR | SH30538.03 | |
| IV butterfly infusion set with 12" tubing and 25G needle | Thermo Fisher Scientific | 26708 | |
| Kimwipes | Kimberly Clark | 34155 | Lint-free wipe |
| LB Agar | Thermo Fisher Scientific | BP1425-500 | LB agar pre-mix for selective media |
| McPherson Vannas iris scissor | Integra LifeSciences | 360-215 | |
| Mineral oil | Sigma Life Science | 69794-500ML | |
| NEB Stable Competent E. coli | NEB | C3040I | |
| NucleoSpin Gel and PCR Clean-Up | Takara | 740609.5 | Kit for enzymatic reaction cleanup and gel extraction |
| OCT medium | VWR | 25608-930 | |
| Orbital shaker | Cole Parmer | 60-100 | |
| Paraformaldehyde | Sigma-Aldrich | 158127-500G | |
| PCR strip tubes 0.2 mL | VWR | 490003-692 | |
| Peristaltic pump | Gilson | F155005 | |
| Phosphate buffered saline (PBS) 10x | Thermo Fisher Scientific | 70011044 | |
| Phusion Hot Start II High Fidelity DNA Polymerase | Thermo Fisher Scientific | F549L | |
| Powdered milk | Sunny Select | ||
| ProLong Gold Antifade Mountant | Thermo Fisher Scientific | P36934 | |
| QIAquick PCR Purification Kit | QIAGEN | 28106 | |
| rCutSmart Buffer | NEB | B6004S | Buffer for restriction digest with PacI, AscI, and XmaI |
| Restriction enzyme: AscI | NEB | R0558L | |
| Restriction enzyme: PacI | NEB | R0547L | |
| Restriction enzyme: XmaI | NEB | R0180L | |
| SOC outgrowth medium | NEB | B0920S | Recovery medium after transformation |
| Sucrose (RNase/DNase free) | Millipore Sigma | 033522.5KG | |
| TAE buffer | Apex | 20-194 | |
| Transfer tubing | Gilson | F1179941 | For peristaltic pump |
| Triton X100 | Sigma-Aldrich | X100-100ML | |
| Wizard Plus SV Minipreps DNA Purification System | Thermo Fisher Scientific | A1460 | Plasmid mini prep kit |
References
- Pennacchio, L. A., et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 444 (7118), 499-502 (2006).
- Nord, A. S., et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell. 155 (7), 1521-1531 (2013).
- Finucane, H. K., et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nature Genetics. 47 (11), 1228-1235 (2015).
- Nord, A. S., West, A. E. Neurobiological functions of transcriptional enhancers. Nature Neuroscience. 23 (1), 5-14 (2020).
- Hallikas, O., et al. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell. 124 (1), 47-59 (2006).
- Su, J., Teichmann, S. A., Down, T. A. Assessing computational methods of cis-regulatory module prediction. PLoS Computational Biology. 6 (12), 1001020 (2010).
- Visel, A., et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 457 (7231), 854-858 (2009).
- Kvon, E. Z., et al. Genome-scale functional characterization of Drosophila developmental enhancers in vivo. Nature. 512 (7512), 91-95 (2014).
- Yáñez-Cuna, J. O., et al. Dissection of thousands of cell type-specific enhancers identifies dinucleotide repeat motifs as general enhancer features. Genome Research. 24 (7), 1147-1156 (2014).
- Zincarelli, C., Soltys, S., Rengo, G., Rabinowitz, J. E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Molecular Therapy. 16 (6), 1073-1080 (2008).
- Gisselbrecht, S. S., et al. Highly parallel assays of tissue-specific enhancers in whole Drosophila embryos. Nature Methods. 10 (8), 774-780 (2013).
- Lambert, J. T., et al. Parallel functional testing identifies enhancers active in early postnatal mouse brain. eLife. 10, 69479 (2021).
- Chen, Y. -. J. J., et al. Use of “MGE Enhancers” for labeling and selection of embryonic stem cell-derived medial ganglionic eminence (MGE) progenitors and neurons. PLoS ONE. 8, 61956 (2013).
- Graybuck, L. T., et al. Enhancer viruses and a transgenic platform for combinatorial cell subclass-specific labeling. bioRxiv. , 525014 (2019).
- Dimidschstein, J., et al. A viral strategy for targeting and manipulating interneurons across vertebrate species. Nature Neuroscience. 19 (12), 1743-1749 (2016).
- Rubin, A. N., et al. Regulatory elements inserted into AAVs confer preferential activity in cortical interneurons. eNeuro. 7, (2020).
- Hindorff, L. A., et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences of the United States of America. 106 (23), 9362-9367 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Ripke, S., et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 511 (7510), 421-427 (2014).
- Nott, A., et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science. 366 (6469), 1134-1139 (2019).
- Lagunas, T., et al. A Cre-dependent massively parallel reporter assay allows for cell-type specific assessment of the functional effects of genetic variants in vivo. bioRxiv. , (2021).
- DeBoer, E., Antoniou, M., Mignotte, V., Wall, L., Grosveld, F. The human beta-globin promoter; nuclear protein factors and erythroid specific induction of transcription. The EMBO journal. 7 (13), 4203-4212 (1988).
- Kothary, R., et al. Inducible expression of an hsp68-lacZ hybrid gene in transgenic mice. Development. 105 (4), 707-714 (1989).
- Niwa, H., Yamamura, K., Miyazaki, J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 108 (2), 193-199 (1991).
- Kim, D. W., Uetsuki, T., Kaziro, Y., Yamaguchi, N., Sugano, S. Use of the human elongation factor 1α promoter as a versatile and efficient expression system. Gene. 91 (2), 217-223 (1990).
- Banerji, J., Rusconi, S., Schaffner, W. Expression of a β-globin gene is enhanced by remote SV40 DNA sequences. Cell. 27 (2), 299-308 (1981).
- Patwardhan, R. P., et al. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nature Biotechnology. 27 (12), 1173-1175 (2009).
- Arnold, C. D., et al. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 339 (6123), 1074-1077 (2013).
- Melnikov, A., et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nature Biotechnology. 30, 271-277 (2012).
- Kim, J. -. Y., Grunke, S. D., Levites, Y., Golde, T. E., Jankowsky, J. L. Intracerebroventricular viral injection of the neonatal mouse brain for persistent and widespread neuronal transduction. Journal of Visualized Experiments: JoVE. (91), e51863 (2014).
- Liu, D., Zhu, M., Zhang, Y., Diao, Y. Crossing the blood-brain barrier with AAV vectors. Metabolic Brain Disease. 36 (1), 45-52 (2021).
- Prabhakar, S., Lule, S., Da Hora, C. C., Breakefield, X. O., Cheah, P. S. AAV9 transduction mediated by systemic delivery of vector via retro-orbital injection in newborn, neonatal and juvenile mice. Experimental Animals. , (2021).
- Gruntman, A. M., Su, L., Flotte, T. R. Retro-orbital venous sinus delivery of rAAV9 mediates high-level transduction of brain and retina compared with temporal vein delivery in neonatal mouse pups. Human Gene Therapy. 28 (3), 228-230 (2017).
- Inoue, F., Ahituv, N. Decoding enhancers using massively parallel reporter assays. Genomics. 106 (3), 159-164 (2015).
- Gordon, M. G., et al. lentiMPRA and MPRAflow for high-throughput functional characterization of gene regulatory elements. Nature Protocols. 15 (8), 2387-2412 (2020).
- Klein, J. C., et al. A systematic evaluation of the design and context dependencies of massively parallel reporter assays. Nature Methods. 17 (11), 1083-1091 (2020).
- Creyghton, M. P., et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences of the United States of America. 107 (50), 21931-21936 (2010).
- Song, L., Crawford, G. E. DNase-seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harbor Protocols. 2010 (2), 5384 (2010).
- King, D. M., et al. Synthetic and genomic regulatory elements reveal aspects of Cis-regulatory grammar in mouse embryonic stem cells. eLife. 9, 41279 (2020).
- Amit, R., Garcia, H. G., Phillips, R., Fraser, S. E. Building enhancers from the ground up: a synthetic biology approach. Cell. 146, 105-118 (2011).
- Fan, M., Tsai, J., Chen, B., Fan, K., LaBaer, J. A central repository for published plasmids. Science. 307, 1877 (2005).
- Sanger, F., Coulson, A. R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. Journal of Molecular Biology. 94, 441-448 (1975).
- Broussard, G. J., Unger, E. K., Liang, R., McGrew, B. P., Tian, L., Parrot, S., Denoroy, L. L. glutamate with genetically encoded fluorescent sensors. Biochemical Approaches for Glutamatergic Neurotransmission. 130, 117-153 (2018).
- Chen, Y. H., Keiser, M. S., Davidson, B. L. Adeno-associated virus production, purification, and titering. Current Protocols in Mouse Biology. 8, 56 (2018).
- He, C. X., Arroyo, E. D., Cantu, D. A., Goel, A., Portera-Cailliau, C. A Versatile Method for Viral Transfection of Calcium Indicators in the Neonatal Mouse Brain. Frontiers in Neural Circuits. 12, 56 (2018).
- Chen, S. -. Y., Kuo, H. -. Y., Liu, F. -. C. Stereotaxic surgery for genetic manipulation in striatal cells of neonatal mouse brains. Journal of Visualized Experiments: JoVE. (137), e57270 (2018).
- Brenowitz, S. D., Regehr, W. G. Presynaptic imaging of projection fibers by in vivo injection of dextran-conjugated calcium indicators. Cold Spring Harbor Protocols. 2012, (2012).
- Bosch, D., Asede, D., Ehrlich, I. Ex vivo optogenetic dissection of fear circuits in brain slices. Journal of Visualized Experiments: JoVE. (110), e53628 (2016).
- Green, E. K., et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Molecular Psychiatry. 15 (10), 1016-1022 (2010).
- Takahashi, S., Glatt, S. J., Uchiyama, M., Faraone, S. V., Tsuang, M. T. Meta-analysis of data from the Psychiatric Genomics Consortium and additional samples supports association of CACNA1C with risk for schizophrenia. Schizophrenia Research. 168 (1-2), 429-433 (2015).
- McCarty, D. M. Self-complementary AAV vectors; advances and applications. Molecular Therapy. 16 (10), 1648-1656 (2008).
- Haery, L., et al. Adeno-associated virus technologies and methods for targeted neuronal manipulation. Frontiers in Neuroanatomy. 13, 93 (2019).
- Rabani, M., Pieper, L., Chew, G. -. L., Schier, A. F. A massively parallel reporter assay of 3′ UTR sequences identifies in vivo rules for mrna degradation. Molecular Cell. 68, 1083-1094 (2017).
- Kim, Y. -. S., et al. Correcting signal biases and detecting regulatory elements in STARR-seq data. Genome Research. 31, 877-889 (2021).
- Lee, D., et al. STARRPeaker: uniform processing and accurate identification of STARR-seq active regions. Genome Biology. 21, 298 (2020).
- Doni Jayavelu, N., Jajodia, A., Mishra, A., Hawkins, R. D. Candidate silencer elements for the human and mouse genomes. Nature Communications. 11, 1061 (2020).
- Singh, G., et al. A flexible repertoire of transcription factor binding sites and a diversity threshold determines enhancer activity in embryonic stem cells. Genome Research. 31, 564-575 (2021).
- Wang, X., et al. High-resolution genome-wide functional dissection of transcriptional regulatory regions and nucleotides in human. Nature Communications. 9, 5380 (2018).
- Peng, T., et al. STARR-seq identifies active, chromatin-masked, and dormant enhancers in pluripotent mouse embryonic stem cells. Genome Biology. 21, 243 (2020).
- Nair, R. R., Blankvoort, S., Lagartos, M. J., Kentros, C. Enhancer-driven gene expression (EDGE) enables the generation of viral vectors specific to neuronal subtypes. iScience. 23, 100888 (2020).
- Russell, S., et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65 -mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. The Lancet. 390, 849-860 (2017).
- Gaudet, D., et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Therapy. 20, 361-369 (2013).
- Nguyen, G. N., et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nature Biotechnology. 39, 47-55 (2021).
- Nayak, S., Herzog, R. W. Progress and prospects: Immune responses to viral vectors. Gene Therapy. 17 (3), 295-304 (2010).
- Visel, A., et al. A high-resolution enhancer atlas of the developing telencephalon. Cell. 152 (4), 895-908 (2013).
- Shen, S. Q., et al. Massively parallel cis-regulatory analysis in the mammalian central nervous system. Genome Research. 26 (2), 238-255 (2016).
- Shen, S. Q., et al. A candidate causal variant underlying both higher intelligence and increased risk of bipolar disorder. bioRxiv. , (2019).
- Hrvatin, S., et al. A scalable platform for the development of cell-type-specific viral drivers. eLife. 8, 48089 (2019).
