As a representative example, we have described in vivo observation of the EBP comets in the steady-state and regenerating axons of the PLM neurons. PLM neurons are located in the tail region of the worm with a long anterior process that forms a synapse and a short posterior process. PLM neurons grow in the anterior-posterior direction close to the epidermis and are responsible for the gentle touch sensation in the worms. Due to their simplified structure, and amenability to imaging and microsurgery, PLM neurons have been extensively investigated for their microtubule cytoskeleton29, axonal transport30,31, and regeneration19,32, neuronal polarity24,33,34, behavior and aging35,36 and many other neuronal processes. We used PLM neurons to assess microtubule dynamics and orientation in vivo in a transgenic expressing EBP-2::GFP under the mec-4 promoter.
For checking the steady-state dynamics of the microtubules, we picked the worms expressing the Pmec-4-EBP-2::GFP reporter. The worms were mounted in 0.1 µm polystyrene beads on 10% agarose pads. The coverslip was added gently, and the worms were imaged on the spinning disk microscope. The PLM neurons were centered and focused on 63x and imaged with a frame rate of 3.3 frames per second. The anterior process of the PLM showed a majority of the comets moving away from the cell body whereas the posterior process showed a bidirectional movement of the comets (Figure 1). Based on the direction of the comets, the anterior process of the PLM contains unipolar plus-end-out microtubules while the posterior process has mixed polarity (plus and minus-end-out) of microtubules (Figure 2)24.
As an application of the assay, we explored the microtubule dynamics and orientation in the regenerating axons of PLM neurons. Previous studies have established methods of laser mediate axotomy using various types of lasers including, UV, nanosecond, picosecond, and femtosecond lasers31,37,38,39,40,41,42,43. For this study, we used a femtosecond laser to sever the anterior process of the PLM neurons43. Following the injury, the worms were recovered onto seeded NGM plates. The worms were then imaged at multiple time points after injury for the observation of EBP-2::GFP comets. At 2 h after injury, a severed axon was visible however, the comets were drastically reduced in number as compared to an uninjured axon. At 11 h after injury, the axons have elicited a regeneration response in the form of axonal regrowth where a large number of EBP-2::GFP comets were observed (Figure 3).
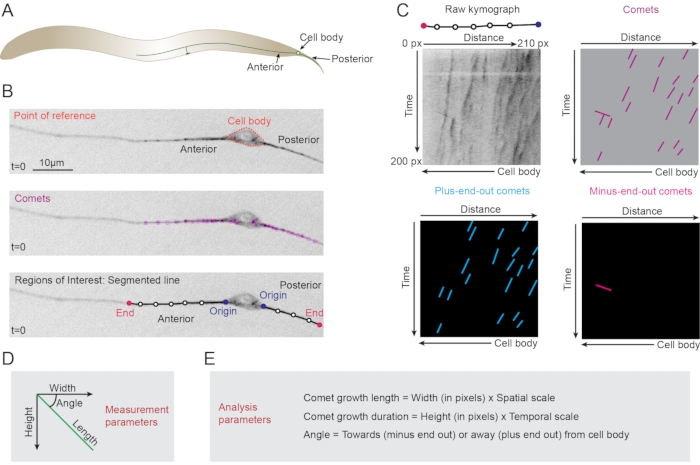
Figure 1: Observation and analysis of EBP comets. (A) Schematic of the PLM neuron showing its position with respect to the worm anatomy. (B) The point of reference is selected as per the neuron type. In this case, the cell body of the PLM neuron serves as the reference point for the regions of interest in anterior or posterior processes. Comets (magenta circles) are visible as punctae in the cell body, anterior and posterior processes of the PLM neuron. A segmented line tracing the neurite of interest (anterior process) is drawn to extract a kymograph. (C) A typical ROI can be converted into a kymograph which is a distance-time image with diagonal traces representing the moving comets. With respect to the cell body, the plus-end-out comets are traced in cyan whereas minus-end-out comets are traced in pink. For spatial reference, the segmented ROI is represented on top of the raw kymograph. (D) Measurement parameters include the width, height, angle, and length of the trace that can be translated into microtubule parameters. (E) Analysis parameters are extracted from the measurement parameters of width, height, and angle of the traces. Please click here to view a larger version of this figure.

Figure 2: Steady-state dynamics of the microtubules in the PLM axon. (A) Snapshots of a representative time series of EBP-2::GFP comets in the PLM neurons showing some of the EBP-2::GFP comets (colored arrows) in the anterior and posterior processes of the PLM neuron. (B) Two regions in the anterior process (ROI-A, and ROI-B) are converted to kymographs in which the traces corresponding to the comets marked in 2(A). The traces have been color and number coded with respect to the moving comets in 2(A). Lower graphic panel represents the observed traces distinguished based on their direction of the movement. Plus end out comets are marked in cyan whereas minus end out comets are marked in pink. For reference, location of the cell body is depicted at the bottom. (C) ROI-C in the posterior process is converted into the kymographs with traces corresponding to the moving comets in posterior process in the Figure 2(A). Rightmost panel represents the plus end and minus end out traces with respect to the position of the cell body marked at the bottom. Notice that most of the traces in the axonal regions (ROI-A, and ROI-B) are moving away from the cell body representing plus-end-out microtubules (cyan traces). On the contrary, in the posterior process (ROI-C), the traces are bidirectional representing the mixed polarity with plus (cyan traces) and minus-end-out (pink traces) microtubules. Please click here to view a larger version of this figure.
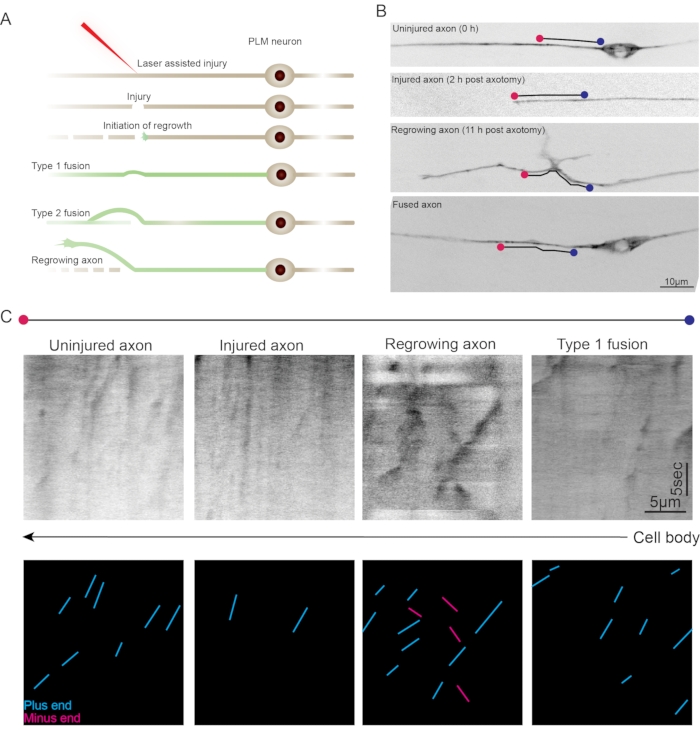
Figure 3: Microtubules during axonal regeneration in PLM neurons. (A) Schematic representation of the axotomy procedure using a femtosecond laser. The laser creates an injury followed by the initiation of the regrowth. Major regeneration responses are scored as Type 1 fusion, Type 2 fusion, and Regrowing axons. (B) Representative images of PLM neurons expressing EBP-2::GFP in the uninjured (0 h), axotomized (2 h post axotomy), and regrowth conditions (11 h post axotomy). The ROI traced for the kymographs have been marked on the images. (C) Representative kymographs of axonal regions traced on the uninjured, injured, regrowing, and Type 1 fused axons. Notice that the number of EBP-2::GFP comets significantly decreased in the injured axons followed by their robust movement (increased growth length and duration) in the regrowing axon. Individual traces can be quantified and classified as per their direction (cyan and pink traces). Please click here to view a larger version of this figure.
