The success of submitochondrial fractionation protocol depends on obtaining highly purified intact mitochondria. For this, it is essential that during the yeast cell lysis, the intactness of the organelles remains almost totally preserved. This is achieved by using a cell lysis protocol that combines the enzymatic digestion of the cell wall followed by physical disruption of the plasma membrane by using a Dounce homogenizer. The mitochondrial contents are then collected by differential centrifugation. This subcellular fractionation yields an enriched mitochondrial fraction, as confirmed by the presence of high levels of porin (Por1), a mitochondrial marker protein (Figure 1, lane 2). However, this is a crude mitochondria fraction, which contains substantial amounts of other cellular compartments, including endoplasmic reticulum, vacuole, cytosol, and endosome (Figure 1, lane 2). These contaminations may introduce artifacts in some applications, such as submitochondrial protein localization experiments. To decrease the amount of these contaminations, the crude mitochondrial fraction is further purified on sucrose density gradient centrifugation. This additional purification step generates a highly pure mitochondrial fraction, as evidenced by a significant reduction in the contents of the protein markers for other cellular compartments (Figure 1, lane 3).
In order to determine the submitochondrial localization of proteins, the highly purified mitochondria are further fractionated into their subcompartments (Figure 2A). This protocol involves the conversion of mitochondria into mitoplasts by hypotonic osmotic shock. In this process, intact mitochondria are incubated in a hypoosmotic buffer resulting in swelling of the organelle. During swelling, the outer mitochondrial membrane is selectively ruptured by osmotic unbalance and the intermembrane space protein content is released into the supernatant. All this procedure is performed in the presence or absence of proteinase K. As a consequence of the outer membrane disruption, the protease gains access to intermembrane space protein content and promotes the degradation of the corresponding proteins. In contrast, the protein content of the mitochondrial matrix remains protected from attack of the protease due to the integrity of the inner mitochondrial membrane. After these treatments, the protein content of the different samples is evaluated by SDS-PAGE and western blot analyzes.
The efficient conversion of mitochondria to mitoplasts by osmotic shock (swelling) can be monitored in two ways: (1) disappearance of the soluble intermembrane space marker protein (e.g., cytochrome Cyt. b2) in the pellet fraction from mitoplasts with its concomitant appearance in the supernatant fraction (Figure 2B, compare lane 3 with lane 7); (2) selective degradation of the inner-membrane marker protein facing the intermembrane space (e.g., ScoI) by proteinase K only in mitoplasts (Figure 2B, lane 4). In addition, the protection of the markers Cyt. b2 and ScoI against proteinase K degradation in the pellet fraction from mitochondria are used to confirm the integrity of the outer mitochondrial membrane (Figure 2B, lane 2). On the other hand, the integrity of the inner mitochondrial membrane is confirmed by the protection of the matrix soluble protein marker α-KGD against proteinase K degradation (Figure 2B, lane 4). To determine the submitochondrial localization of a protein of interest, simply compare its western blot profile with the profiles of these standards with known localization.
In the case of the protein depicted in Figure 2B (Prx1), its western blot profile is indicative of a protein with dual mitochondrial localization: intermembrane space and matrix. At first glance, its fractionation profile is similar to α-KGD, indicating a matrix localization. However, its presence in the supernatant of mitoplasts also indicates an intermembrane space localization. The fractionation profiles of protein markers described above eliminate the possible artifacts associated with the integrity of the mitochondrial preparation and corroborate the dual localization of Prx119.
To investigate the topology of proteins on mitochondrial membranes, mitochondria are submitted to two additional treatments: sonication and carbonate extraction (Figure 3A). While sonication releases only soluble proteins into the supernatant fraction13, alkaline extraction with sodium carbonate additionally solubilizes peripherally membrane-associated proteins13,20. In both the treatments, integral membrane proteins remain in the pellet fraction13. These assumptions are confirmed by western blot analysis of the pellet and supernatant fractions from both treatments (Figure 3B). An integral mitochondrial membrane protein (e.g., porin Por1) is expected to be found totally in the pellet fraction, even after the alkali treatment with sodium carbonate (Figure 3B, lanes 3 and 5). On the other hand, a soluble matrix protein (e.g., α-KGD) is expected to be completely solubilized in both the treatments (Figure 3B, lanes 2 and 4). The significant retention of α-KGD in the pellet from sonication (Figure 3B, lane 3, SMP fraction) might be due to slight variations in the sonication parameters that can affect the formation of the so-called submitochondrial particles, which are efficiently sedimented by ultracentrifugation. The behavior of these proteins with known solubility profiles are then used to determine the mitochondrial solubility of a protein of interest. In the case of Prx1, its western blot profile is suggestive of a protein associated with the membrane periphery and alkaline treatment induces its solubilization (Figure 3B, lane 4).
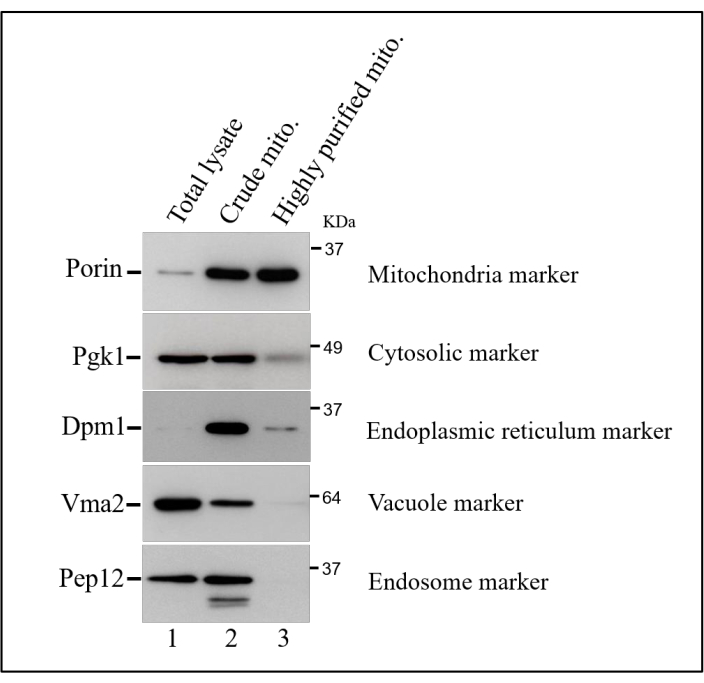
Figure 1: Isolation of highly purified mitochondria. Western blot analysis of total lysate fraction (lane 1), a crude mitochondrial fraction (lane 2), and highly purified mitochondrial fraction (lane 3). The fractions were separated by SDS-PAGE on a 12% polyacrylamide gel, transferred to nitrocellulose and probed with antibodies raised against markers for distinct cellular compartments as described on the right side of the gel. This figure has been modified from reference19. Please click here to view a larger version of this figure.
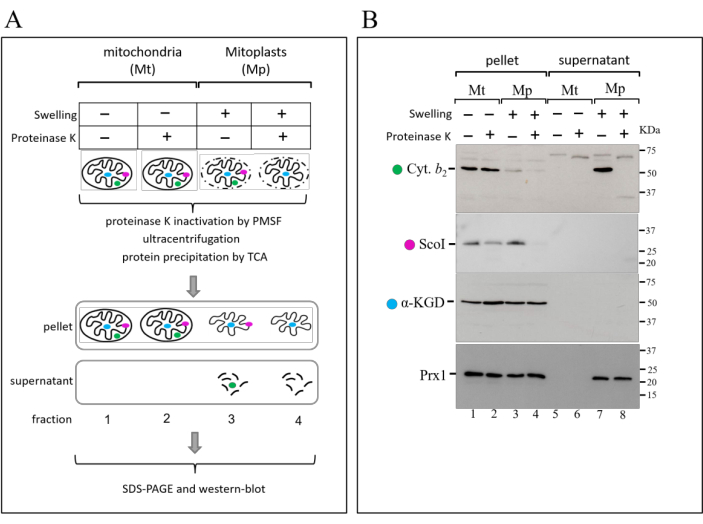
Figure 2: Submitochondrial fractionation protocol by hypotonic swelling in the presence of proteinase K. (A) Schematic representation of submitochondrial fractionation protocol. Highly purified mitochondria are separately subjected to isotonic or hypotonic treatments (swelling) in the presence (+) or absence (-) of proteinase K. After treatment, the activity of proteinase K is inhibited by the addition of PMSF, and mitochondria and mitoplasts are recovered by centrifugation. The protein content from the resulting pellet and supernatant fractions is precipitated by TCA, and then analyzed by SDS-PAGE and western blot. The color spheres represent submitochondrial protein markers for: a soluble intermembrane space protein (green), an inner membrane protein that faces the intermembrane space (pink), and a soluble matrix protein (light blue). (B) Western blot analysis of pellet and supernatant fractions from submitochondrial fractionation protocol. The fractions were separated by SDS-PAGE on a 12% polyacrylamide gel, transferred to nitrocellulose and probed with antibodies raised against markers for distinct submitochondrial compartments as depicted in A. See text for more details. This figure has been modified from19. Please click here to view a larger version of this figure.
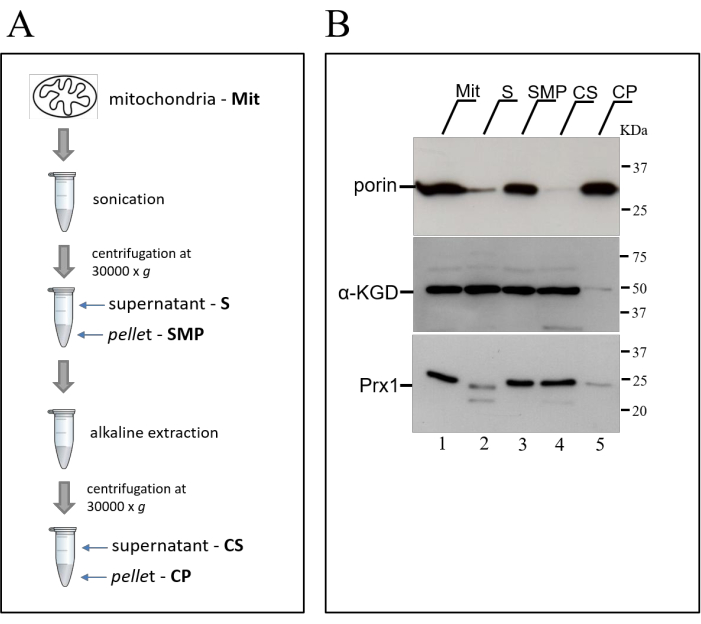
Figure 3: Submitochondrial fractionation protocol by sonication and carbonate extraction. (A) Schematic representation of the protocol used to determine the solubility and membrane topology of mitochondrial proteins. Mitochondria are initially sonicated and centrifuged, resulting in a soluble protein fraction (S), and the compartmentalized membranous product, called submitochondrial particle (SMP). The pellet from the sonication step is subsequently submitted to an alkaline treatment with sodium carbonate (Na2CO3) and centrifuged, resulting in carbonate supernatant (CS) and carbonate-precipitated fractions (CP). (B) Western blot analysis of pellet and supernatant fractions from sonication and carbonate extraction protocol. The fractions were separated by SDS-PAGE on a 12% polyacrylamide gel, transferred to nitrocellulose and probed with antibodies raised against protein markers showing distinct levels of solubility. See text for more details. This figure has been modified from19. Please click here to view a larger version of this figure.
| Solution | Components | Comments | |
| YPD medium | 1% (w/v) yeast extract, 2% (w/v) peptone, 2% (w/v) glucose | Dissolve 10 g Bacto Yeast extract, 20 g Bacto Peptone and 20 g glucose in 900 m of distilled water added. Fill up to 1000 ml and sterilize by autoclaving. | |
| YPGal medium | 1% (w/v) yeast extract, 2% (w/v) peptone, 2% (w/v) galactose | Dissolve 10 g Bacto Yeast extract, 20 g Bacto Peptone and 20 g galactose in 900 m of distilled water added. Fill up to 1000 ml and sterilize by autoclaving. | |
| DTT buffer | 100 mM Tris-H2SO4 (pH 9.4), 10 mM dithiothreitol (DTT) | To make 15 ml: mix 1.5 ml of 1 M Tris-H2SO4, pH 9.4 with 150 µL of 1M DTT prewarmed at 30 °C. Volume to 15 ml with ddH2O Prepare freshly prior to use |
|
| Zymolyase buffer | 20 mM potassium phosphate buffer (pH 7.4), 1.2 M sorbitol | To make 100 ml: mix 2 ml of 1 M potassium phosphate buffer (pH 7.4) with 60 ml of 2 M sorbitol Volume to 15 ml with ddH2O Dissolve the powder (3 mg per gram wet weight) of Zymolyase-20T from Arthrobacter luteus (MP Biomedicals, Irvine, CA) in the buffer just before use |
|
| Homogenization buffer | 10 mM Tris-HCl (pH 7.4), 0.6 M sorbitol, 1 mM EDTA, 0.2% (w/v) bovine serum albumin (BSA), 1 mM phenylmethylsulfonyl fluoride (PMSF) | To make 250 ml: mix 2.5 ml of 1 M Tris-HCl buffer (pH 7.4) with 75 ml of 2 M sorbitol, 500 μL of 500 mM EDTA and 0.5 g of BSA (essentially fatty acid-free) Volume to 250 ml with ddH2O Pre-cool at 4°C Add PMSF and BSA in the buffer just before use |
|
| SEM buffer | 10 mM MOPS-KOH (pH 7.2), 250 mM sucrose, 1 mM EDTA | To make 250 ml: mix 2.5 ml of 1 M MOPS-KOH buffer (pH 7.2) with 31.25 ml of 2 M sucrose and 500 μL of 500 mM EDTA Volume to 250 ml with ddH2O Pre-cool at 4°C just before use. |
|
| EM buffer | 10 mM MOPS-KOH (pH 7.2), 1 mM EDTA | To make 250 ml: mix 2.5 ml of 1 M MOPS-KOH buffer (pH 7.2) with 500 μL of 500 mM EDTA Volume to 250 ml with ddH2O Pre-cool at 4°C just before use |
|
| sample buffer | 2% (w/v) sodium dodecylsulfate (SDS), 50 mM DTT, 10% (v/v) glycerol, 0.02% bromophenol blue, 60 mM Tris-HCl (pH 6.8), |
||
Table 1: Media, solutions, and buffers.
| Reagent | 1 | 2 | 3 | 4 | |
| Mitochondria (10 mg/mL) | 40 µL | 40 µL | 40 µL | 40 µL | |
| SEM buffer | 360 µL | 360 µL | – | – | |
| EM buffer | – | – | 360 µL | 360 µL | |
| Proteinase K (10 mg/mL) | – | 4 µL | – | 4 µL | |
Table 2: Pipetting scheme to perform hypotonic swelling.
