As described above, the strains used for the single-cell phenotypic analysis of persister cells were characterized in MOPS glycerol 0.4% medium. The monitoring of the OD600nm over time showed no difference between the wt and hupA-mCherry strains (Figure 1). This indicates that the expression of the HU-mCherry fusion protein did not impact growth in these conditions. The bacterial cells of both strains initially inoculated at an OD600 nm of 0.01 reached the exponential phase ±8 h after inoculation.
The MIC of OFX was determined by standardized methods (here, serial agar dilution)24. MIC is defined as the minimal concentration where no visible growth is detected. The MIC of OFX for both strains was determined to be 0.06 µg·mL−1, indicating that the hupA-mCherry fusion had no effect on the sensitivity to OFX in comparison with the isogenic wt strain (Figure 2).
We further determined the effect of a lethal OFX treatment (83-fold MIC) on the viability of both strains used in this study. As the viable cell count decreases over time with OFX exposure, the dilutions of bacterial cultures need to be adjusted appropriately to reach 30 to 300 colonies per plate. To determine the appropriate dilutions over time, a spot assay was performed, where 10 µL from 0 to 10−7 serial 10-fold dilutions were placed on square Petri dishes using a multi-channel pipette. The appropriate dilutions were those where isolated clones were visible (e.g., at t0 = 10−5, t1h = 10−4/10−3, t4h = 10−2/10−1) (Figure 3).
While the spot assay is an easy method to gain insights into the kinetics of OFX-mediated killing, it fails to accurately determine the killing dynamics. When the viability of exponentially growing cells treated with OFX was monitored by the time-kill assay, a typical biphasic curve was observed (Figure 4). The first slope of the curve reflects the rapid killing of the non-persister population (red dashed line). In the conditions tested here, up to 99.9% of the cells were unable to form colonies after 3 h in the presence of OFX. This first phase of killing is followed by a second phase, showing a slower killing rate (blue dashed line), which reveals the presence of drug-tolerant persister cells. In the conditions tested, the persister phase started around 3 h after the OFX addition, highlighting the necessity to expose the cells to OFX for longer than 3 h to investigate the persister phenotypes. Importantly, the time-kill curve shows that the hupA-mCherry fusion protein had no effect on the time-kill kinetics. The strain encoding the translational fluorescent fusion can, therefore, be used to monitor the persister cells using fluorescence microscopy.
We further went on to investigate the persistence phenomenon at the single-cell level. To do so, the hupA-mCherry strain was introduced into a microfluidic plate, which allowed for the change of medium conditions (here, growth, treatment, and recovery) while performing time-lapse microscopy on a given ROI. During the first step of the microfluidic experiment, the cells introduced into the microfluidic device were perfused with growth medium (MOPS glycerol 0.4%) and divided with a generation time of ~2 h (Figure 5 and Figure 6). This first phase of growth indicates that cells were viable and actively dividing before the OFX treatment.
After this first phase of growth, the cells were perfused with growth medium supplemented with 5 µg·mL−1 OFX for 6 h. As soon as the antibiotic reached the cells, cell division was blocked (Figure 5 and Figure 6). After 6 h of OFX treatment, the cells were perfused with fresh medium. While the vast majority of the cells were unable to resume growth (Figure 5 and Figure 6), a small subpopulation of bacteria was capable of elongating and generating filamentous cells25. These cells, which were able to divide and generate viable daughter cells after the OFX treatment, can be defined as the persister cells.
As this setup allows for the visualization of the persister cells before, during, and after treatment, it not only provides information about the persister phenotype during the recovery phase but also about the physiological state of the persister cells before the treatment (Figure 6). In the conditions tested, the persister cells divided similarly to non-persister cells prior to the OFX treatment, indicating that the observed persister cells did not originate from a dormant subpopulation (Figure 6)25.
The cell length analysis of persister cells during the recovery phase revealed that each filament had a specific rate of elongation. The cell length reached by each persister before the first division differed from one persister to another. Similarly, the timing of the first division event was highly heterogeneous (Figure 6). The dividing persister filament generated multiple daughter cells, which started to grow and divide, for the most part, similarly to untreated cells (Figure 7). The successive division of the filament then resulted in a progressive decrease in the cell length, ultimately giving rise to daughter cells with similar cell length to before the OFX treatment (Figure 6 and Figure 7B). The vast majority of the cells were unable to induce filamentation after OFX removal. This large cell population corresponds to the dead cells (Figure 5 and Figure 6).
The fluorescent fusion of the nucleoid-associated protein HU allows for the visualization of the dynamics of the nucleoid22. The analysis of the total fluorescence intensity of HU-mCherry within the cell can be used as a proxy for the DNA content22,25. During the growth phase (before OFX treatment), the total mCherry fluorescence intensity varied, reflecting the dynamics of chromosome replication and segregation during the cell cycle (Figure 8). After OFX addition, the mCherry fluorescence increased at the mid-cell, indicative of nucleoid compaction, which has been shown to be induced by the formation of double-strand DNA breaks28 (Figure 5). Double-strand DNA breaks are a consequence of the mechanism of action of OFX, which corrupts the type II topoisomerases DNA-gyrase and topoisomerase IV29,30. In E. coli, DNA-gyrase is the primary target of OFX29,30. By binding its target at a critical step of the double-strand passage mechanism, OFX inhibits the relegation of the cleaved DNA strands, ultimately leading to the release of double-strand DNA breaks30. As described above, the persister cells to OFX treatment started to filament during recovery25 (Figure 6). The increase in cell length correlated with an increase in the total mCherry fluorescence intensity, which reflects replication restart and an increase in the nucleoid abundance in the filament25 (Figure 7a and Figure 8). For dead cells, the total mCherry fluorescence intensity remained stable during the treatment and during the recovery phase, indicating that these cells were unable to replicate their chromosomes after OFX removal (Figure 8). Microfluidic video (Video 1) of E. coli HU-mCherry cells before, during, and after ofloxacin treatment is also shown.
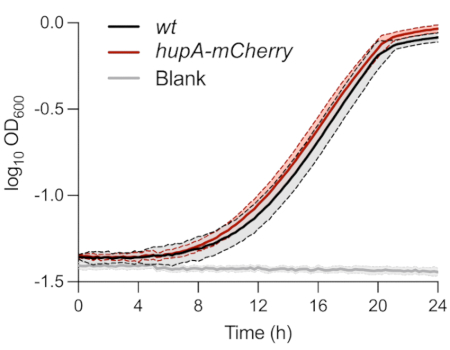
Figure 1: Growth monitoring of wt and hupA-mCherry E. coli strains. Optical density monitoring (OD600 nm) of wt (black) and hupA-mCherry (red). The shades and dashed lines indicate the standard deviations of the biological triplicates. Please click here to view a larger version of this figure.
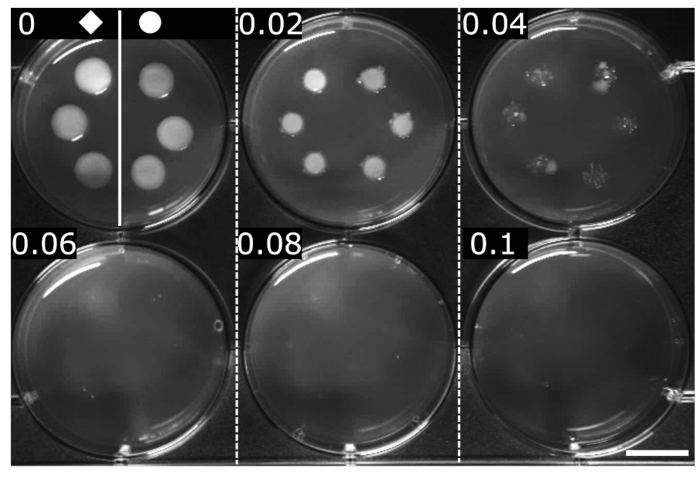
Figure 2: Determination of the MIC of OFX for the wt and hupA-mCherry E. coli strains. The wt (♦) and hupA-mCherry (●) were grown in LB medium, and 2 µL were spotted on serial dilutions of OFX-containing LB agar (concentration indicated in each panel in µg·mL−1). Growth inhibition is visible at a minimum of 0.06 µg·mL−1. The figure is a representative experiment of biological triplicates. Scale bar = 1 cm. Please click here to view a larger version of this figure.
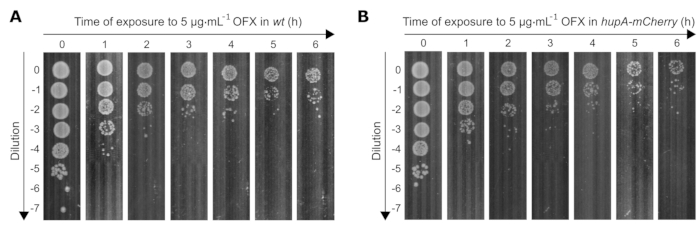
Figure 3: Spot assay of wt and hupA-mCherry E. coli strains upon exposure to OFX. The (A) wt and (B) hupA-mCherry strains were grown in MOPS glycerol 0.4% as described in the protocol (section 3), and the exponentially growing cells (OD600 nm = 0.3) were treated with 5 µg·mL−1 OFX. T0 corresponds to the time point before the addition of OFX. T1, T2, T3, T4, T5, and T6 correspond to 1-6 h after the OFX addition. The figure is a representative experiment of biological triplicates. Please click here to view a larger version of this figure.
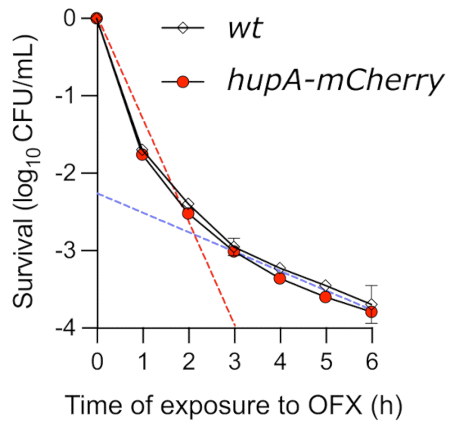
Figure 4: Time-kill assay of wt and hupA-mCherry E. coli strains upon exposure to OFX. The wt (♦) and hupA-mCherry (●) strains were grown in MOPS glycerol 0.4% as described in the protocol (section 4), and the exponentially growing cells (OD600 nm = 0.3) were treated with 5 µg·mL−1 OFX. The dashed lines indicate the first "rapid" (red) killing phase and the second "slow" (blue) killing phase, corresponding to the sensitive and persistent subpopulations (obtained by linear regression between T0 and T2, as well as between T3 and T6, respectively). The error bars indicate the standard deviations of the biological triplicates. Please click here to view a larger version of this figure.
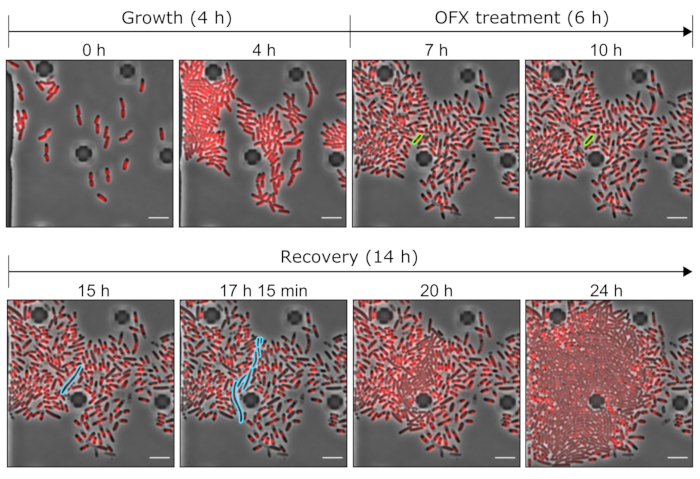
Figure 5: Representative images of the OFX persister and dead cells using microfluidic tools. Representative microscopy images showing the relevant time points of the microfluidic experiment performed with the hupA-mCherry strain (phase contrast in grey, HU-mCherry signal in red). The cells expressing the tagged hupA-mCherry were grown in a microfluidic plate (here, 4 h), followed by an OFX challenge (5 µg·mL−1). After 6 h in the presence of OFX, the cells were perfused with fresh medium, allowing the persister cells to recover. The persister cell and its progeny cells during the OFX treatment and after OFX removal are highlighted in green and blue, respectively. The corresponding time points are indicated on each panel. Scale bar = 5 µm. Please click here to view a larger version of this figure.
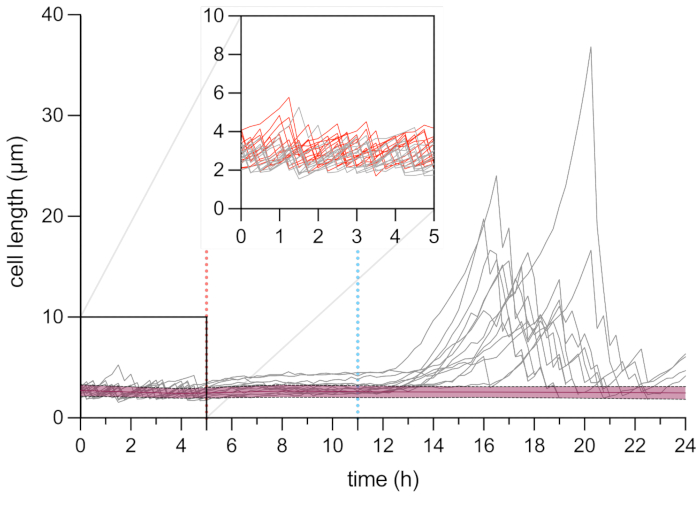
Figure 6: Microscopy time-lapse analysis of the length of the persister and dead cells. Cell length analysis of dead cells (in red, n = 109) and persister cells (in grey, n = 13). The start of the OFX treatment (5 µg·mL−1) is indicated by the red dashed line (5 h), and the OFX removal is indicated by the blue dashed line. The inset corresponds to the growth phase before OFX addition. The experiments were performed in triplicate. The shades and dashed lines indicate the standard deviations for the dead cell population (n = 109). Please click here to view a larger version of this figure.
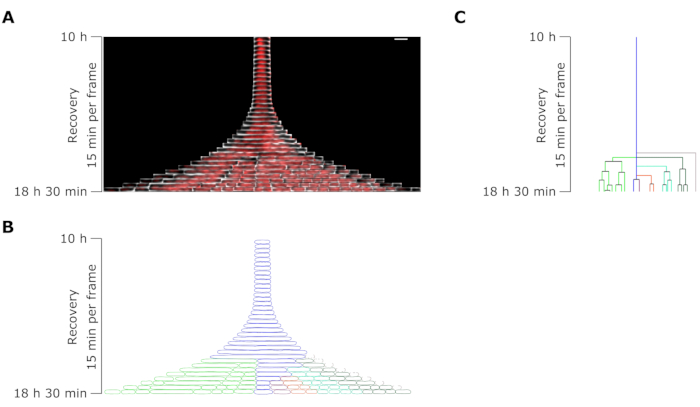
Figure 7: Microscopy time-lapse analysis of a representative persister to OFX. (A) Kymograph of a representative OFX persister and its daughter cells generated by filament divisions during 8.5 h after OFX removal (18.5 h after the beginning of the microfluidic experiment, comprising 4 h of growth, 6 h of 5 µg·mL−1 OFX treatment, and 8.5 h of recovery after OFX removal). One frame corresponds to 15 min. Scale bar = 5 µm. (B) Mask generated from the persister kymograph in A. The monitored persister cell is indicated with a blue outline, and the daughter cells are highlighted in distinct colors. (C) Schematic representation of the persister cell lineage generated from B. The color coding is identical to B. Please click here to view a larger version of this figure.
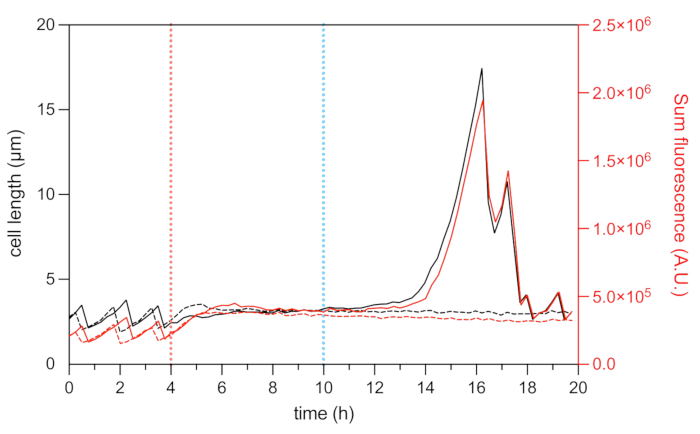
Figure 8: Cell length and mCherry fluorescence analysis of representative persister and dead cells. Analysis of the cell length (left axis) and the total HU-mCherry fluorescence intensity (right axis, shown in arbitrary units) of a representative persister (solid black and red lines) and a representative dead cell (dashed black and red line) during the microfluidic time-lapse experiment. The start of the OFX treatment (5 µg·mL−1) is indicated by the red dashed line, and the OFX removal by the blue dashed line. Please click here to view a larger version of this figure.
Video 1: Microfluidic video of E. coli HU-mCherry cells before, during, and after ofloxacin treatment. Microfluidic time-lapse imaging showing HU-mCherry cells. The cells were grown for 4 h in MOPS glycerol 0.4%. After 6 h of OFX treatment (5 µg·mL−1), the antibiotic-free medium was perfused in the microfluidic plate to allow the persister cells to recover. Scale bar = 5 µm. Time (in min) is indicated. The growth and recovery phases are indicated by "MOPS- Gly. 0.4%" and the OFX treatment by "OFX 5 µg/mL". Please click here to download this Video.
| 10x MOPS | |||
| Stock solution | Volume of stock solution for 1 L of 10x MOPS | Final concentration in 10x MOPS base | |
| MOPS acid | 1 M (adjusted to pH 7.4 using KOH) | 400 mL | 0.4 M |
| Tricine | 1 M (adjusted to pH 7.4 using KOH) | 40 mL | 0.04 M |
| FeSO4.7H2O | 0.01 M | 10 mL | 0.0001 M |
| NH4Cl | 1.9 M | 50 mL | 0.095 M |
| K2SO4 | 0.276 M | 10 mL | 0.00276 M |
| CaCl2.2H2O | 0.0005 M | 10 mL | 0.000005 M |
| MgCl2.6H2O | 0.528 M | 10 mL | 0.00528 M |
| NaCl | add directly 29.2 g | 0.5 M | |
| Distilled water | 460 mL | ||
| Micronutriments 1000x (see Table 2) | 10 mL | ||
Table 1: Composition of 10x MOPS.
| Micronutriments 1000x | ||
| Concentration in Micronutriments 1000x stock solution | Final concentration in 10x MOPS base | |
| (NH4)6Mo7O24.4H2O | 0.000003 M | 0.00000003 M |
| H3BO3 | 0.0004 M | 0.000004 M |
| CoCl2.6H2O | 0.00003 M | 0.0000003 M |
| CuSO4.5H2O | 0.00001 M | 0.0000001 M |
| MnCl2.4H2O | 0.00008 M | 0.0000008 M |
| ZnSO.7H2O | 0.00001 M | 0.0000001M |
Table 2: Composition of 1,000x micronutrients.
| MOPS glucose 0.4% or MOPS glycerol 0.4% | |||
| Stock solution | Volume for 1 L MOPS glucose 0.4 % or MOPS glycerol 0.4 % | Final concentration in MOPS glucose 0.4% or MOPS glycerl 0.4% | |
| 10x MOPS | see Table 1 | 100 mL | |
| K2HPO4 | 0.132 M | 10 mL | 0.00132 M |
| Glucose (for MOPS glucose 0.4%) | 20% (20 g in 100 mL distilled water) | 20 mL | 0.40% |
| Glycerol (for MOPS glycerol 0.4%) | ≤99% | 4 mL | 0.40% |
| Distilled water | 870 mL for MOPS glucose 0.4% or 886 mL for MOPS glycerol 0.4% | ||
Table 3: Composition of MOPS glucose 0.4% and MOPS glycerol 0.4%.
