To illustrate the use of CorrelationCalculator, we constructed a partial correlation network using a subset of the metabolomics data from the KORA population study described in Krumsiek et al.24. The dataset contained 151 metabolites and 240 samples. Figure 1 shows the resulting partial correlation network that was visualized in Cytoscape. The network contains 148 nodes and 272 edges. The color of the nodes represents metabolites that belong to different chemical classes, while the edges represent the adjusted p-value of the partial correlation coefficients (adjusted p-value < 0.05) . Notably, despite not using any prior information CorrelationCalculator was able to group together chemically related metabolites. For example, phosphatidylcholines and lysophosphatidylcholines are closely connected in the network. Visualizing metabolite changes in the context of this type of network can facilitate hypothesis generation, help plan future experiments and enable manuscript preparation. To illustrate a potential workflow utilizing a partial correlation metabolite network, we performed consensus network clustering as described in Ma et al.22, resulting in the identification of 9 subnetworks or metabolic modules. These modules had a good agreement with the chemical classes, i.e., metabolites belonging to the same chemical class tended to be part of the same metabolic module. The user can access the clustering tool clusterNet at https://github.com/Karnovsky-Lab/clusterNet.
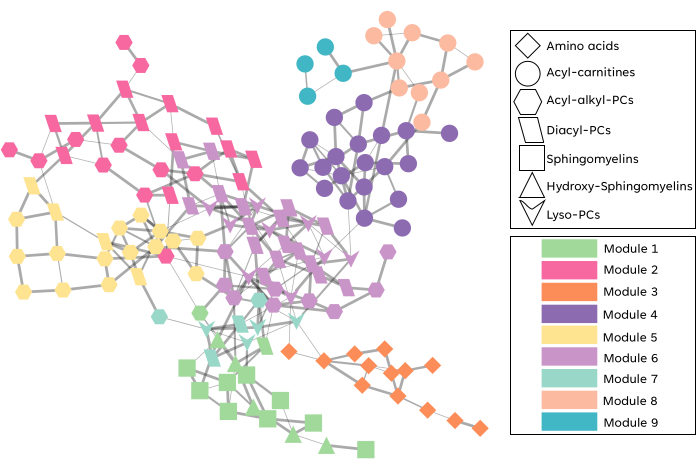
Figure 1: Representative example of a CorrelationCalculator network. The network was constructed from a subset of the KORA population study metabolomics data24 consisting of 151 metabolites across 240 subjects. The nodes represent metabolites, and the edges connecting them are weighted by the adjusted p-value of partial correlation coefficients (adjusted p-value < 0.05). The shape of the nodes represents different metabolic classes, and the color represents metabolic modules obtained by clustering the network using the consensus clustering method. Please click here to view a larger version of this figure.
We illustrate the application of Filigree by analyzing a dataset from a mouse model of type I diabetes (T1D)25,26. Plasma metabolite measurements from T1D and non-diabetic (NOD) mice were used to generate a differential partial correlation network (Figure 2). Notably, we observe a higher degree of network connectivity in the non-diabetic group. The next steps of the analysis identified twelve metabolic modules, nine of which were significantly different between T1D and non-diabetic mice (FDR < 0.05). We refer the reader to the original publication for further insights into biological conclusions that can be drawn from this analysis21.
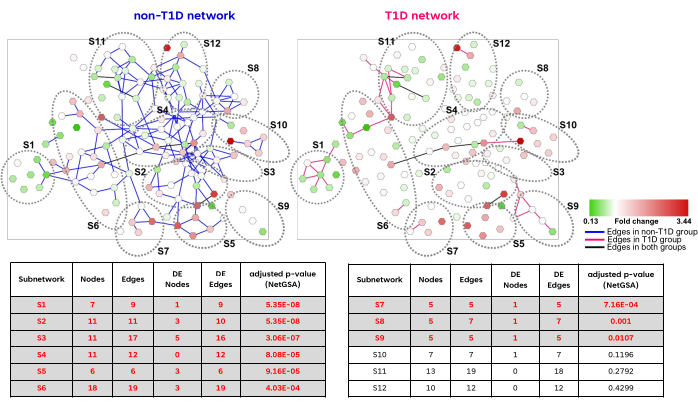
Figure 2: Representative example of a Filigree network. The differential network was constructed utilizing the levels of 163 metabolites from 71 mice (30 T1D and 41 non-T1D)25,26. Differential edges between T1D and non-T1D groups are indicated in pink and blue, respectively. The nodes are colored based on the fold change. The table shows the enrichment results produced by Filigree. Nine out of the twelve identified subnetworks were significantly different between T1D and non-T1D (adjusted p-value < 0.05). Please click here to view a larger version of this figure.
Supplementary Figure 1: CorrCalc_InputTab. Screenshot of the Correlation Calculator's Input tab. Please click here to download this File.
Supplementary Figure 2: CorrCalc_DataNormTab. Screenshot of the Correlation Calculator's Data Normalization tab. Log-2 Transform Data and Autoscale Data are checked. Please click here to download this File.
Supplementary Figure 3: CorrCalc_DataAnalTab. Screenshot of the Correlation Calculator's Data Analysis tab showing filtering to Pearson's Correlation of 0-0.8. In addition, the DSPC Method has been selected. Please click here to download this File.
Supplementary Figure 4: Filigree_DataTab. Screenshot of Filigree's Data tab. Columns, rows, and groups have been specified. The Calculate Feature Groups method has been selected with a feature reduction of 1.25 feature-to-sample ratio. Please click here to download this File.
Supplementary Figure 5: Filigree_AnalysisTab. Screenshot of Filigree's Analysis tab showing the progress of the different analysis components. Please click here to download this File.
Supplementary Figure 6: Filigree_Subnetwork1. A subnetwork generated from Filigree. Node color represents up/down-regulation, and color opacity represents higher/lower fold change. The edge color represents the differential status between groups. Please click here to download this File.
Supplementary Figure 7: Filigree_Subnetwork_SampleGroup. Subnetwork separated by group. The left network represents diabetic samples, and the right network represents non-diabetic samples. Node color represents expression level proportional to the group mean. Please click here to download this File.
