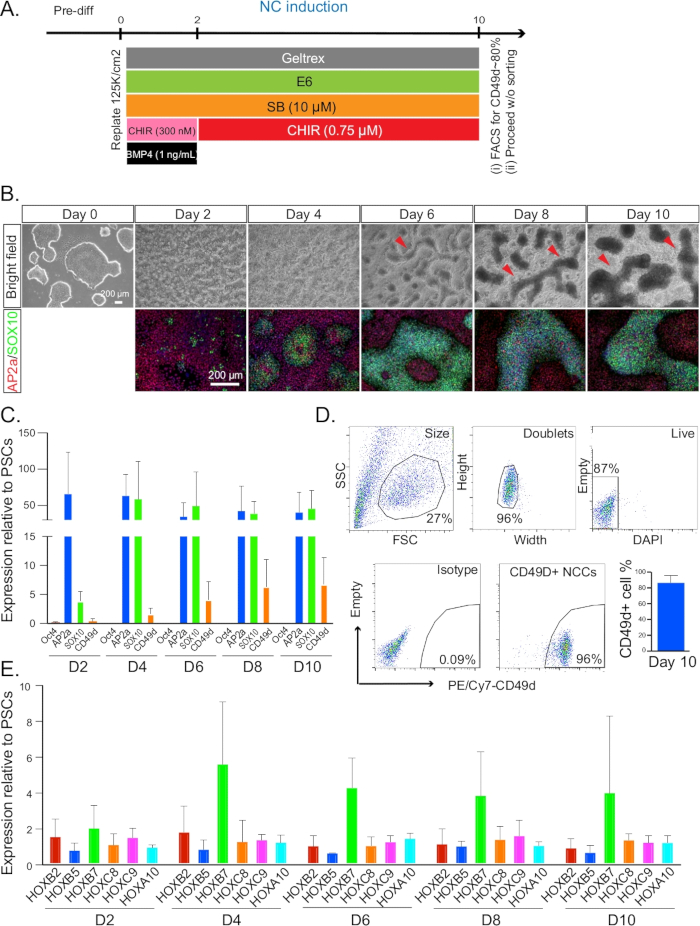
Figure 1: NC induction. (A) Timeline and treatments for NC induction from day 0 to day 10. (B) The morphology and formation of NC ridges were monitored every 2 days by bright field microscopy. Cells at each time point after day 2 were co-stained for AP2A (red) and SOX10 (green). All immunofluorescence pictures were counterstained with DAPI. Red arrows indicate the structures of ridges. (C) qRT-PCR analysis for the expression profile of NC markers from day 2–10. (D) Representative plot of FACS sorting on day 10 for CD49D+ NC populations (left). A typical gating strategy and isotype control is indicated in the first four plots. Quantification of CD49D+ cell percentage on day 10 was also performed. (E) qRT-PCR analysis for the expression profile of HOX genes from day 2–10. NC = neural crest. Error bars stem from data of n ≥ 3 biological repeats, defined as independent differentiation experiments done on separate days from PSC cultures that were at least one split apart.
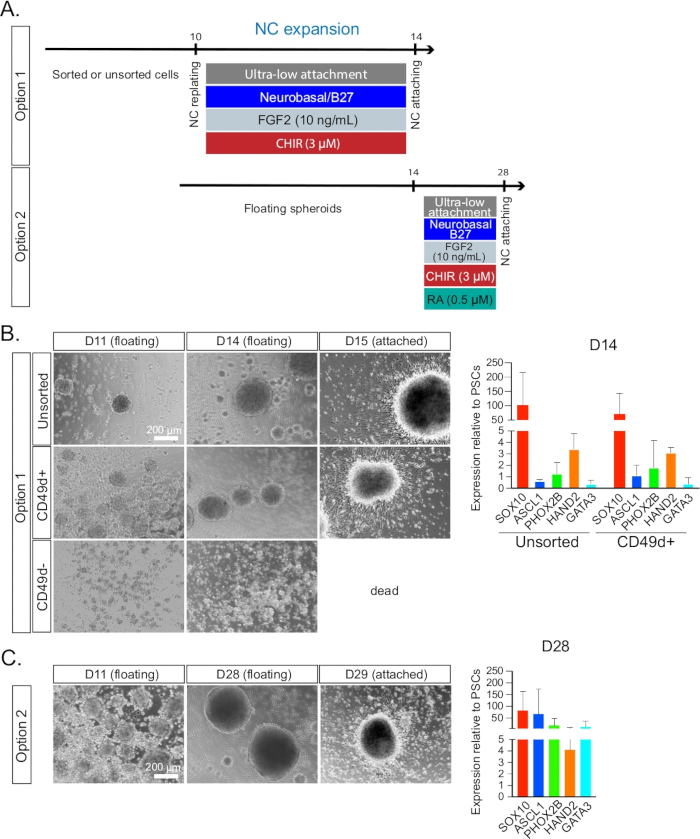
Figure 2: Neural crest maintenance and expansion. (A) Timeline and treatments for NC expansion during day 10 to day 14 culture for option 1 or day 10 to day 28 culture for option 2. (B) The NC spheroid formation was monitored by bright field microscopy from day 11 (1 day after spheroid formation) to day 14 (i.e., the day of plating). Unsorted, CD49D+, and CD49D– populations were compared. The plated cells on day 15 are also shown. Cells failed to form spheroids properly in the CD49D– group and died after plating. Day 14 cells were examined by qRT-PCR analysis (right) for the expression profile of symN progenitors. Unsorted and CD49D+ populations were compared (n ≥ 3). (C) NC spheroids could be maintained for up to 2 weeks. Spheroids were monitored by bright field microscopy from day 15 to day 28 (the day of landing). The plated cells on day 29 are also shown. Day 28 spheroids were examined by qRT-PCR analysis for the expression profile of symN progenitors (right). Unsorted and CD49D+ populations were pooled (n ≥ 3).
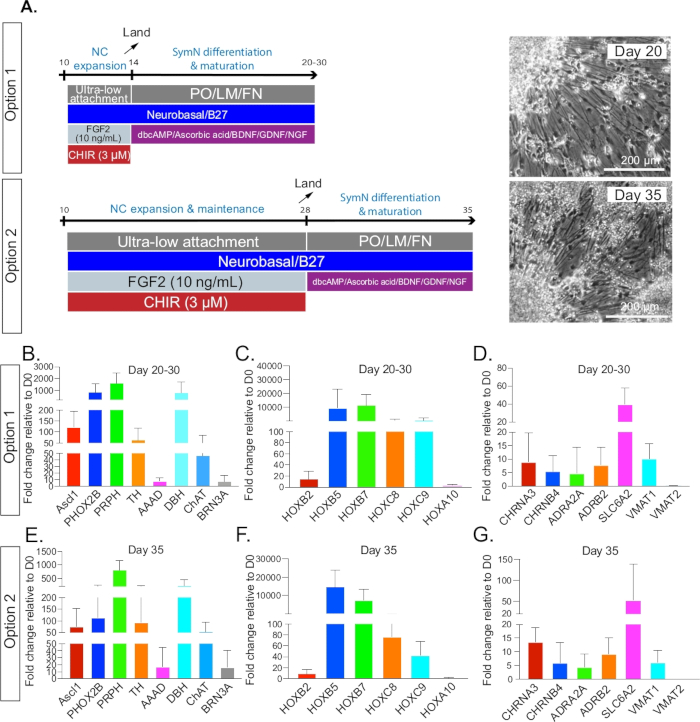
Figure 3: SymN differentiation and maturation. (A) Timeline and treatments after plating on day 14 for option 1 or day 28 for option 2. Bright field microscopy photos (right) show symNs on day 20 and day 35, respectively (1 week after plating for both options). (B–D) Option 1 qRT-PCR analysis for the expression profile of symN properties between days 20–30. Unsorted and CD49D+ populations were pooled. (E–G) Option 2 qRT-PCR analysis for the expression profile of symN properties after day 35. Unsorted and CD49D+ populations were pooled (n ≥ 3).
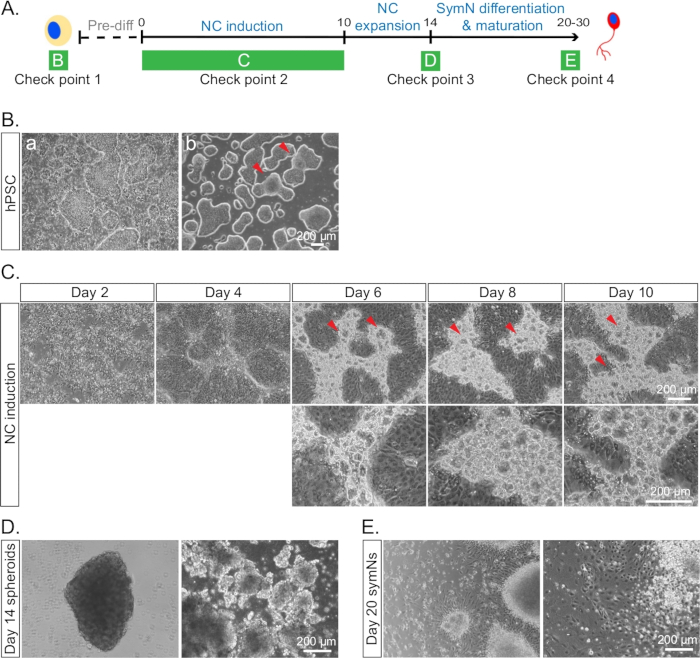
Figure 4: Example of cells under conditions that are inappropriate to proceed with the differentiation. (A) Timeline of the differentiation with each check point for cell morphologies indicated as in B–E. (B) hPSCs (a) with healthy colonies and a lot of differentiated cells, and (b) merged and differentiated borders between some colonies on day 0. Red arrows indicate the merged areas. (C) NCCs at day 10 with bubble-like blisters in the ridges. Red arrows indicate the blisters in the top row. The bottom row represents a higher magnification. We have not been able to identify the cell identity of the blisters. However, presence of more blisters seems to correlate with lower SOX10/CD49D expression. (D) Irregular looking spheroids lacking smooth edges and round shape on day 14. (E) Unhealthy and dying symNs on day 20.
