1. Sample preparation
- Use fresh, flash-frozen or RNAlater-treated tissue from mice as starting material. Blot away any excess preservative with a Kimwipe before proceeding.
- Remove all tendon and fascia around the muscle and mince the tissue with single edged razor blades for 2 min on chilled glass plates in a cold room (4°C) until the tissue is well minced or powdery. Collect the sample in a pre-weighed microfuge tube and determine the weight of the tissue. Use no more than 10 mg of the tissue as your starting material.
- Add 45 μL extraction buffer (8 M urea, 50 mM DTT, 4% CHAPS, 0.2% carrier ampholytes 5/8, 0.0002% bromophenol blue) per 1 mg minced tissue, at room temperature. If the volume is greater than 350 μL, add 350 μL initially and homogenize (step 1.4), then add the rest of the buffer to minimize splashing.
- Homogenize the tissue seven times with a motorized pestle (Kontes Corp) for 15 seconds at 4°C, with two minutes cooling on ice between each homogenization.
- Centrifuge the samples at 15,600 x g for 30 minutes at 4°C. Save the supernatant and measure its volume. The cell debris pellet is discarded.
- Precipitate the proteins in the supernatant with ice-cold reagent-grade acetone (3 times v:v ratio). Vortex the tubes for 15 seconds. Centrifuge for 15,600 x g for 10 seconds to form pellets, and resuspend in extraction buffer with the Kontes motor and polypropylene stirring rods (BioSpec Prod)
- Repeat step 1.6.
- Either proceed with protein concentration estimation of the samples or store them at -20°C.
2. Protein concentration estimation
Use the Lowry assay7 or the BCA assay8 (Pierce); aim for about 4.5 mg ml-1.
3. Isoelectric focusing
- Remove a ReadyStrip IPG strip (11 cm, pH 5-8, Bio-Rad) from -20°C and allow it to equilibrate at RT for 10-15 min.
- Vortex the sample and pipette 600-800 μg (total volume 200 μL) of protein sample in a straight line at the back edge of a channel in the sample rehydration tray, leaving about 1 cm at each end.
- Using forceps, peel off the plastic cover of the IPG strip – make sure to handle only the ends of the strip and avoid any contact with the gel. Note the basic end of the strip and position it at the left side of the tray. Place the IPG strip gel-side down on top of the sample, avoid trapping bubbles underneath. Allow it to rehydrate for one hour.
- Overlay with 2.5 mL mineral oil (BioRad). Cover the tray with the lid, and leave to rehydrate overnight at room temperature.
- Place a paper wick at both ends of the focusing tray channel and wet each one with 10 μL Millipore water.
- Take out IPG strip and hold vertically for about 10 seconds the drain the oil. Blot the plastic backing with a Kimwipe.
- Place the IPG strip gel side down and with basic end to the left in the focusing tray. Cover the strip with 2.5 mL mineral oil.
- Place the tray into the PROTEAN IEF cell (Bio-Rad) and program a 3-step protocol (Table 1) at default cell temperature of 20°C, a maximum current of 50 μA/strip, and no rehydration time.
Voltage Time Volt-Hrs Ramp Step 1 250 20 min Linear Step 2 8,000 2.5 hr Linear Step 3 8,000 40,000 Rapid Total 6.5 hr 40,000
Table 1. Three-step protocol of PROTEAN IEF cell for 11 cm IPG strips. - Take the IPG strips out of the IEF cell using forceps. Blot the plastic backing with a Kimwipe and place the IPG strip gel side up in a clean rehydration tray. You can cover the rehydration tray and wrap it with plastic wrap and store at – 80°C or proceed.
4. SDS polyacrylamide gel electrophoresis
- Transfer the strip (gel side up) into an 11 cm equilibration tray.
- Add 4 mL of reduction buffer (6 M urea, 2% SDS, 0.05 M Tris/HCl pH 8.8, 20% glycerol, 2% DTT) to the channel and equilibrate the strip for 10 min with mild shaking.
- Remove the reduction buffer, add 4 mL of alkylation buffer (6 M urea, 2% SDS, 0.05 M Tris/HCl pH 8.8, 20% glycerol, 2.5% iodoacetamide) and equilibrate the strip for 10 minutes with mild shaking.
- Rinse the IPG strip by dipping the strip briefly in 1 X SDS running buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.2).
- Lay the strip gel side up onto the back plate of the 11 cm Criterion pre-cast 10.5%-14% Tris-HCl SDS polyacrylamide gel (Bio-Rad) and push it gently in so that it makes full contact with the SDS gel; make sure no bubbles are trapped between the two gel surfaces.
- Overlay the IPG strip with 1 mL of molten agarose solution (0.5% agarose, 25 mM Tris, 192 mM glycine, 0.1% SDS, bromophenol blue) and let the agarose solidify for 5 minutes.
- Load 5 μL of protein marker 10-225 kDa (USB, P/N: 76740) into the single well as molecular weight standards.
- Run the gel in SDS buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.2) at 200 V at room temperature or in a cold room (4°C) using the bromophenol blue dye front migration to monitor the electrophoresis.
- Remove the gel from the plastic cast and stain overnight by shaking gently in Coomassie Brilliant Blue R-250 stain (45.5% methanol, 45.5% dH2O, 9.0% acetic acid, 0.25% Coomassie Brilliant Blue R-250).
- Shake the gel in Destain 1 solution (45.5% methanol, 45.5% dH2O, 9.0% acetic acid) twice for 3 hours each and in Destain 2 (5% methanol, 90% dH2O, 5% acetic acid) overnight. Store the gel in 7% acetic acid for analysis.
5. Gel imaging and analysis
- Capture images of the gels using the Quantity One software program that operates the VersaDoc imaging system (BioRad). Crop the image areas uniformly for all gels in your experiment.
- Load the cropped images on the PDQuest 8.0 software program to create an experiment set. Follow the Experiment setup wizard to define the parameters for spot detection and spot matching across the gels. Inspect and refine the spot matching results manually if needed.
- Perform quantitative and statistical analysis of the gels to detect matched protein spots with two-fold or higher statistically significant differences in expression between the two gel groups. Create an analysis set with these spots of interest and cut them out using an EXQuest spot cutter (BioRad) for trypsin digestion and LC-MS-based protein identification.
6. In-gel tryptic digestion
For this step a modified Pierce In-Gel Tryptic Digest Kit (#89871X, Pierce) protocol is used.
- Add 200 μL of destaining solution (25 mM ammonium bicarbonate, 50% acetonitrile) to the gel plugs. Vortex and incubate the samples at 37°C for 30 minutes with shaking. Carefully remove and discard the solution.
- Repeat step 6.1.
- Add 30 μL of freshly prepared reducing buffer (50 mM Tris[2-carboxyethyl] phosphine, 22.5 mM ammonium bicarbonate) to the tubes containing the gel plugs and incubate at 60°C for 10 minutes.
- Allow samples to cool; then remove and discard the reducing buffer.
- Add 30 μL of alkylation buffer (100 mM iodoacetamide, 20 mM ammonium bicarbonate, prepared immediately before use in foil-wrapped tubes. Incubate the samples in the dark at room temperature for 1 hour.
- Remove and discard the alkylation buffer. Wash the gel plugs by adding 200 μL of destaining solution to each tube. Incubate the samples at 37°C for 15 minutes.
- Remove and discard the destaining solution and repeat the wash.
- Add 50 μL of acetonitrile to the gel plugs and incubate them for 10 minutes at room temperature to allow them to shrink dry, then carefully remove the acetonitrile.
- Repeat step 6.8 once more. Gel pieces should look white and small.
- Allow gel pieces to dry in a Centrivap Concentrator (Labconco, Model EX1245) for 10 minutes.
- Swell the gel pieces by adding 10 μL of activated trypsin solution (10 ng/μL trypsin, 25 mM ammonium bicarbonate). Incubate at room temperature for 15 minutes.
- Add 25 μL digestion buffer (25 mM ammonium bicarbonate) to the tubes. Incubate the samples at room temperature overnight with shaking.
- Sonicate the samples for 10 minutes (Branson, bath sonicator model 2510) and spin them down before removing the supernatant to a fresh tube. Save the supernatant.
- To further extract peptides, add 10 μL of 0.1% formic acid and incubate for 5 minutes at room temperature with shaking. Sonicate the samples for 10 minutes, collect the supernatant and combine with saved supernatant in step 6.13.
7. Microscale desalting of peptide extracts
- Remove ammonium bicarbonate and other salts in the peptide extracts with the help of PepClean C-18 Spin Columns (ThermoFisher) according to manufacturer instructions.
- Elute peptides twice with 20 μL 70% acetonitrile, evaporate the samples dry by centrifugation in the Centrivap Concentrator for 1 hour and resuspend the peptides in 10 μL 0.1% formic acid for MS analysis.
8. Protein identification by HPLC-coupled mass spectrometry
- Separate 5 μL of the peptide mixture on a high performance liquid chromatograph (HPLC) over 40 minutes, using a linear 2-50% acetonitrile gradient with 0.2% formic acid on a Pepswift monolithic PS-DVB column (Dionex) coupled to a Nanospray I source onto an ion trap mass spectrometer (LCQ Deca XP Max, Thermo Fisher Scientific) at a flow rate of 400 nL/min at the tip.
- Detect peptide MS1 signals between 400 – 1400 m/z and allow for the 3 most intense ion peaks to be isolated for MS2 sequencing by CID with dynamic exclusion.
- For protein identification, analyze the peptide results via a Sequest mouse reference database search (BioWorks software, Thermo-Fisher Scientific) with a static alkylated cysteine modification and dynamic modifications for phosphorylation (serine, threonine, tyrosine), methylation (histidine), oxidation (methionine), ADP-ribosylation (arginine), and N-terminal acetylation. Apply conservative matching criteria (e.g., peptide tolerance set to 2 AMU and 1.00 fragment tolerance, proteins contain at least 2 peptides passing an Xcorr vs Charge State filter [z=1/x=1.5, z=2/x=2.00, z=3/x=2.50, z=4/x=3.00] and a probability of p < 0.05)5 and manually inspect the MS spectra for confident protein identification.
9. Representative Results:
Male and female unexercised, murine skeletal muscle (biceps brachii) was extracted and separated into a two-dimensional map5, the first level of the muscle comparative proteome (Figure 2). Once visualized using high-resolution digital imaging; about 800 protein spots were detected in each. Using the male proteome map as a baseline, it is clear that there are numerous spots that change in abundance in the female proteome. The spot intensities are measures of the change in the protein amounts (Figure 3). The identities of the protein spots that change more than two-fold (up or down) and are statistically significant (p < 0.05) with an n=5 mice, are determined by amino acid sequence analysis using liquid chromatography-coupled mass spectrometry. These results show that females demonstrate an abundance decrease in sugar energy metabolism enzymes and in creatine kinase enzyme (CK) isoforms, a different energy supply system in muscles. Both humans and animals display higher male serum CK levels at rest and following exercise9,10, but serum CK levels do not necessarily correlate with the amount of myofibrillar disruption11,12. This gender dimorphism in the cellular abundance of CK in murine biceps brachii muscle is a novel finding5 and may answer the physiologically different serum CK levels.
Figures:
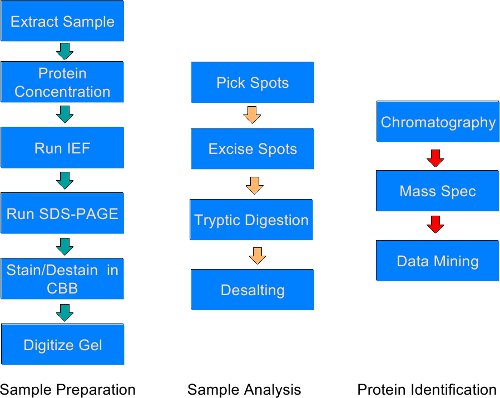
Figure 1. An overall schematic of the protocol for comparative proteomics. The protocol is subdivided into three groups: sample preparation; sample analysis; and, protein identification. The ends of each of these three groups of protocols are also reasonable pausing places, although the best results are garnered if the overall protocol is carried out without stopping.
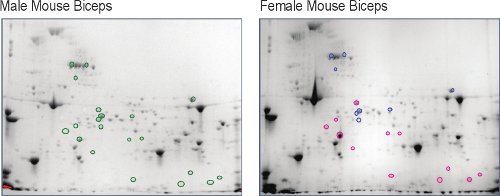
Figure 2. Comparison of female murine biceps brachii protein spots relative to the identical spots in male biceps brachii (green circles o). Spots that increased greater than or equal to two-fold in females are circled red (o) and those that decreased less than or equal to two-fold are circled blue (o).

Figure 3. Gel Spot Intensity Analysis: X-axis, female pre-exercise (control, n=5); Y-axis, female single bout 0 hr time point group (n=5). Regression line: correlation coefficient = 0.919; slope = 0.976; intercept = -0.0293. Spots above the red line and below the blue line change more than +/- two-fold.
