Next generation sequencing of viruses from clinical sources can inform transmission and the epidemiology of infections, as well as help support novel diagnostic, vaccine and therapeutic development. cDNA synthesis using random primers has allowed the detection and assembly of genomes from divergent, co-infecting or even novel viruses1,2. As with other unbiased methods, unwanted contaminants occupy many sequencing reads and negatively impact sequencing results. Host and poly(rA) carrier RNA are contaminants present in many existing viral sample collections.
The protocol describes an efficient and cost-effective way of deep sequencing RNA virus genomes based on unbiased total RNA-seq. The method utilizes an RNase H selective depletion step3 to remove unwanted host ribosomal and carrier RNA. Selective depletion enriches for viral content (Figure 1) and improves the overall quality of sequencing data (Figure 2) from clinical samples. Moreover, tagmentation is applied to the protocol as it significantly reduces library construction time. These methods have been used to rapidly generate large datasets of Ebola and Lassa virus genomes2,4,5 and can be used to study a wide range of RNA viruses. Lastly, the approach is not limited to human samples; the utility of selective depletion was demonstrated on tissue samples collected from Lassa-infected rodents and non-human primate disease models5,6.
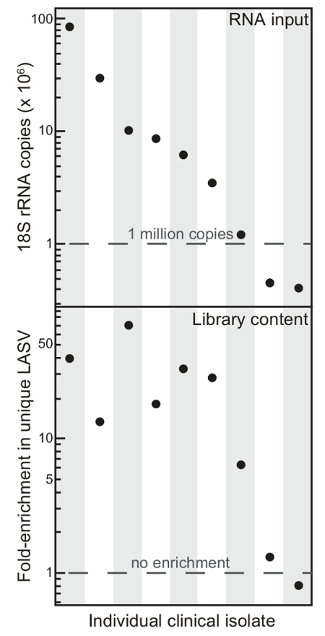
Figure 1. Total RNA Content Reflects Enrichment of Lassa Virus Content Using Selective Depletion. Starting overall content (RNA input) and enrichment of unique Lassa virus (LASV) reads (Library content) upon rRNA depletion from nine different clinical isolates. This figure has been modified from6. Please click here to view a larger version of this figure.
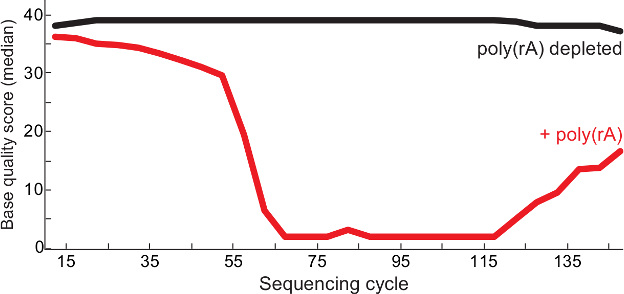
Figure 2. Higher Quality Sequencing After Carrier RNA Depletion. Median base qualities per sequencing cycle of poly(rA)-contaminated Lassa virus libraries (red) and control (no carrier observed in library, black) from QC report 13. Both read 1 and read 2 of paired end reads are merged in the library BAM file and the quality scores are shown at each base. This figure has been modified from6. Please click here to view a larger version of this figure.
The viral RNA-seq protocol details construction of libraries directly from extracted RNA collected from clinical and biological samples. To ensure personal safety, all viral serum, plasma and tissue samples should be inactivated in appropriate buffers prior to RNA extraction. In some inactivation and extraction kits, carrier poly(rA) RNA is included; this will be removed during the initial RNase H selective depletion step. Based on complete recovery, the expected concentration of carrier RNA is 100 ng/µl. In the protocol, 110 ng/µl oligo dT RNA (1.1x carrier concentration) is used for depletion. If poly(rA) carrier is not present in the sample, then oligo(dT) should not be added prior to depletion.
The following protocol is designed for 24 reactions in PCR plate format (up to 250 µl volume). An earlier version of this protocol was reported in Matranga, et al.6.
Ethics statement: Lassa fever patients were recruited for this study using protocols approved by human subjects committees at Tulane University, Harvard University, Broad Institute, Irrua Specialist Teaching Hospital (ISTH), Kenema Government Hospital (KGH), Oyo State Ministry of Health, Ibadan, Nigeria and Sierra Leone Ministry of Health. All patients were treated with a similar standard of care and were offered the drug Ribavirin, whether or not they decided to participate in the study. For Lassa fever (LF) patients, treatment with Ribavirin followed the currently recommended guidelines and was generally offered as soon as LF was strongly suspected.
Due to the severe outbreak for Ebola Virus Disease (EVD), patients could not be consented through our standard protocols. Instead use of clinical excess samples from EVD patients was evaluated and approved by Institutional Review Boards in Sierra Leone and at Harvard University. The Office of the Sierra Leone Ethics and Scientific Review Committee, the Sierra Leone Ministry of Health and Sanitation, and the Harvard Committee on the Use of Human Subjects have granted a waiver of consent to sequence and make publically available viral sequences obtained from patient and contact samples collected during the Ebola outbreak in Sierra Leone. These bodies also granted use of clinical and epidemiological data for de-identified samples collected from all suspected EVD patients receiving care during the outbreak response. The Sierra Leone Ministry of Health and Sanitation also approved shipments of non-infectious, non-biological samples from Sierra Leone to the Broad Institute and Harvard University for genomic studies of outbreak samples.
1. DNase-treatment of Sample RNA (Up to 55 µl Extracted Total RNA, ~4 hr)
- Set up the DNase reaction in a 96-well PCR plate on ice in a biosafety cabinet as described in Table 1, Step 1.1 (total volume, 70 µl/well). Note: A master mix can be prepared.
- Vortex gently and thoroughly, then centrifuge at 280 x g at RT for 1 min.
- Incubate at 37 °C for 30 min.
- Cleanup using RNA Solid Phase Reversible Immobilization (SPRI) beads.
- Warm RNA beads to RT for 30 min.
- Gently shake RNA beads bottle to resuspend any magnetic particles that may have settled. Add 1.8x volume (126 µl) of RNA beads to DNase-treated RNA (70 µl), mix by pipette 10 times and incubate for 5 min at RT (total volume in well, 196 µl).
- Place mixture on the magnetic station. Wait for the solution to clear (5 – 10 min).
- Remove cleared solution while on the station by pipette and discard. While on station, wash beads by covering pellet with 70% ethanol and incubate for 1 min. Remove ethanol with pipette and discard. Repeat for a total of two washes.
Note: Using precisely 70% freshly prepared ethanol is critical, as a higher percentage will result in inefficient washing of smaller-sized molecules, whereas <70% ethanol could cause loss of sample7. - Keep plate on the station and leave open to air-dry. Note: Be sure to allow the beads to dry completely until beads begin to crack.
- Add 55 µl of nuclease-free water to the plate to elute RNA. Remove plate from the station to mix the beads and water by pipetting thoroughly. Note: Alternatively, use less water (≤ 10 µl) in order to concentrate the total RNA.
- Place plate back on the station. Wait until solution clears to transfer by pipette to new screw-cap tube for long-term storage (-80 °C). Place 5 µl RNA in new 96-well PCR plate for depletion (step. 2.4).
- Optional: Save and dilute 1 µl in 19 µl water (1:20) for qRT-PCR of rRNA (e.g., 18S, 28S rRNA) (Table 2) and viral markers5.
2. Selective Depletion of Ribosomal and Carrier RNA from Viral RNA Sample (~4 hr)
- Make 5x hybridization and 10x RNase H reaction buffers, and nuclease-free water with linear acrylamide carrier as described in Table 1.
- Set up hybridization reaction by combining RNA with rRNA depletion oligos (Table 3) and oligo(dT) on ice in a 96-well PCR plate as described in Table 1.
Note: A master mix can be prepared. 50 femtograms (fg) of a unique synthetic RNA (ERCCs8) can be added for tracking both the viral sequencing process and potential index read cross-contamination.- Vortex gently and thoroughly, then centrifuge at 280 x g at RT for 1 min.
Incubate at 95 °C for 2 min, slow ramping to 45 °C at -0.1 °C per sec. Pause the thermocycler at 45 °C.
- Vortex gently and thoroughly, then centrifuge at 280 x g at RT for 1 min.
- Set up RNase H reaction mix on ice as described in Table 1, then preheat at 45 °C for 2 min. Note: A master mix can be prepared.
- Add the pre-heated RNase H mix to the hybridization reaction in plate while keeping the plate in the thermocycler at 45 °C.
- Mix well by gentle pipetting 6 – 8 times. Incubate at 45 °C for another 30 min. Place on ice.
- Set up the DNase reaction mix on ice as described in Table 1. Note: A master mix can be prepared.
- Add to the RNase H reaction in plate, vortex gently and thoroughly, then centrifuge at 280 x g at RT for 1 min. Incubate at 37 °C for 30 min.
- Stop DNase reaction by adding 5 µl 0.5 M EDTA. Vortex gently and thoroughly, then centrifuge at 280 x g at RT for 1 min.
- Cleanup using RNA beads (see step 1.3) using a 1.8x volume (144 µl) beads. Elute in 11 µl of nuclease-free water. Note: For safe cold storage, store depleted RNA samples at -80 °C O/N.
3. cDNA Synthesis (~6 hr)
- Mix rRNA/carrier-depleted RNA with random primers on ice in a 96-well PCR plate as described in Table 1, vortex gently and thoroughly, then centrifuge at 280 x g at RT for 1 min.
- Heat the mixture to 70 °C for 10 min in a thermocycler. Immediately after heat denaturation, place the RNA on ice for 1 – 5 min. Do not allow the RNA to stand (even on ice) for longer than 5 min prior to the first-strand reaction.
- Set up first-strand synthesis reaction mix on ice as described in Table 1.
Note: A master-mix may be prepared.- Add to RNA/random primer mix in plate, vortex gently and thoroughly, then centrifuge at 280 x g at RT for 1 min. Incubate at 22 – 25 °C for 10 min.
- Incubate at 55 °C in an air incubator for 60 min. Place the plate on ice to terminate the reaction. Note: The use of an air incubator is recommended to create gradual warming of the first-strand reaction during which the primers anneal and the first strand begins to elongate.
- Set up second-strand synthesis reaction mix on ice as described in Table 1.
Note: A master-mix may be prepared.- Add to the first-strand synthesis reaction in the plate, vortex gently and thoroughly, then centrifuge at 280 x g at RT for 1 min. Incubate for 2 hr at 16 °C (keep lid at 25 °C). Do not allow the temperature to rise above 16 °C.
- Place the plate on ice, then inactivate reaction by adding 5 µl of 0.5 M EDTA, mix gently and thoroughly, then centrifuge at 280 x g at RT for 1 min.
- Cleanup with DNA beads (see step 1.3 for protocol) using 1.8x volume (153 µl) of beads. Elute in 9 µl of elution buffer (EB). Save 1 µl for quantification. Use 1 ng of cDNA for subsequent steps. If cDNA concentration is too low to detect, use 4 µl of cDNA for tagmentation (see step 4.1).
- For safe cold storage, store double-stranded cDNA at 4 °C O/N or -20 °C for long-term storage.
4. Library Preparation — DNA Library Construction (~4 hr)
- Transfer 4 µl of cDNA to a 96-well plate and save the remaining cDNA for a second attempt if needed.
- Set up the tagmentation reaction on ice as described in Table 1.
Note: A master-mix may be prepared. To reduce background and overall cost, the total volume of the tagmentation reaction is reduced from 20 to 10 µl. As cDNA is the limiting factor, the amount of ATM (i.e., transposome) used in the reaction is also reduced to decrease the number of integration sites.- Add tagmentation mix to cDNA in the plate, vortex gently and thoroughly and centrifuge at 280 x g (at RT) for 1 min. Incubate at 55 °C for 5 min, hold at 10 °C.
- Once at 10 °C, immediately add 2.5 µl of Neutralize Tagment Buffer (NT) to end the reaction. Mix by pipetting up and down, then centrifuge at 280 x g (at RT) for 1 min.
- Incubate at RT for 5 min.
- Set up PCR amplification reaction on ice as described in Table 1.
- Vortex gently and thoroughly, then centrifuge at 280 x g at RT for 1 min.
- Perform PCR on thermocycler using the conditions described in Table 1.
Note: 12 cycles of PCR are suggested for 1 ng of tagmented cDNA; however, viral clinical samples often have undetectable amounts of cDNA. For low amounts of cDNA (<1 ng), use up to 18 cycles of PCR to create enough library for sequencing.
- Library preparation — cleanup and pooling for sequencing
- Bring sample up to 50 µl with EB.
- Cleanup with DNA beads (see step 1.3 for protocol) using 0.6x volume (30 µl) beads. Elute in 15 µl EB.
- Determine concentration of library (Figure 3) by conducting region analysis (150 to 1,000 bp) using bioanalyzer software9, excluding primer dimers (~120 bp) from region analysis. Note: Alternatively, qPCR can be used to quantify libraries 10.
- Pool libraries at the lowest molar concentration of 1 nM or greater. If library is below 1 nM, add a small volume of library to pool (~1x volume of other libraries) to capture sequence information from these libraries.
- Cleanup pool with 0.7x DNA beads as outlined above (see step 2). Elute in 15 µl EB. Note: Volume of beads will depend on the final volume of the pool.
- Analyze pool9. Determine molar concentration by conducting region analysis (150 to 1,000 bp)9. Note: Alternatively, qPCR can be used to quantify library pool 10.
- Load sequencer at 10 pM library concentration to generate 101 bp, paired-end reads with dual barcode reads11.
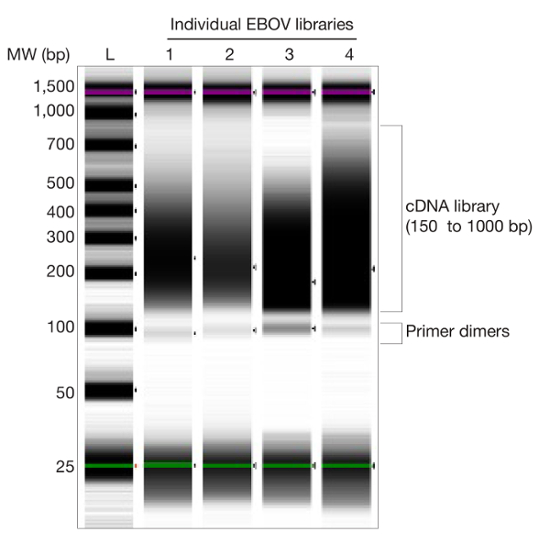
Figure 3. Libraries Constructed from Ebola Virus Clinical Samples. Gel image of 4 representative Ebola virus (EBOV) libraries. Regions of library and primer dimers are shown. Please click here to view a larger version of this figure.
