Formation of the TFO-directed ICL is critical for the plasmid based assays, which are used to interrogate the roles of architectural proteins in ICL processing in human cells. Denaturing agarose gel electrophoresis is a facile way to determine the efficiency of TFO-directed ICL formation. The plasmids harboring TFO-directed ICLs migrate with a slower mobility through the agarose gel matrix (Figure 1A, lane 3) when compared to the un-crosslinked control plasmids (Figure 1A, lane 2). Densitometric quantification of the bands in lanes 2 and 3 (Figure 1A) reveals approximately 70% of the plasmid population migrates through the gel with slowed mobility (identified by a black arrow), indicating TFO-directed ICL formation on those plasmids.
The chromatin immunoprecipitation assays performed on extracts from human U2OS cells transfected with either control plasmid (P) or plasmids containing TFO-directed ICLs (P-ICL) show enrichment of HMGB1 at the P-ICL region compared to the control (P) region, when immunoprecipitated with an anti-HMGB1 antibody (Figure 2, top panel, lanes 2 and 3). These results indicate that HMGB1 associates with the ICLs in human cells. When HMGB1 was depleted from the cells via siRNA treatment, the P-ICL-specific enrichment was diminished (Figure 2, top panel, lanes 4 and 5), as expected. When the same samples were immunoprecipitated using an anti-IgG antibody as an antibody specificity control, no PCR amplification was detected, as expected (Figure 2, top panel, Lanes 6-10). Lanes 10-14 (top panel) in Figure 2 were input samples used for normalization. The top panel shows PCR amplification by the primers near the TFO-directed ICL site and the bottom panel shows PCR amplification when primers distal to the TFO-directed ICL site were used.
HMGB1 is an architectural protein, which binds to distorted DNA with higher affinity than it binds undamaged DNA. The DNA supercoiling assays resolved through two-dimensional agarose gel electrophoresis demonstrate architectural modification of the TFO-directed ICL-containing plasmids (P-ICL) compared to control plasmids (P) by HMGB1 in HeLa cell extracts (Figure 3). The architectural modification induced by HMGB1 preferably on P-ICL-containing substrates is detected as increase in negative supercoiling. Supercoiled plasmids migrate faster through agarose gels relative to relaxed or linear plasmids. The distribution of the topoisomers indicates more supercoiling of the P-ICLs compared to P in the presence of HMBG1 (Figure 3). Negatively supercoiled DNA runs as left-handed arc in the second dimension in the presence of chloroquine. The supercoiling induced by HMGB1 on P-ICL seems to consist mostly of topoisomers that form a left-handed arc (~14 topoisomers were identified on the TFO-directed ICL containing plasmids compared to ~7 topoisomers from the control plasmid), indicating formation of negative supercoils. Altogether, these data suggest that plasmids with TFO-directed ICLs can be used as a tool to study the high-affinity association of the architectural protein HMGB1 with DNA lesions in human cells through chromatin immunoprecipitation assays. In addition, specific architectural modifications of the P-ICL substrates by HMGB1 can also be studied using supercoiling assays and two-dimensional agarose gels.
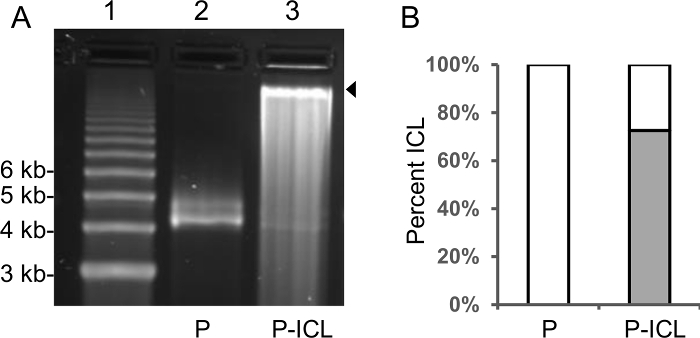
Figure 1: Alkaline agarose gel electrophoresis assay to quantify TFO-directed ICL formation efficiency on the plasmid pSupFG1. (A) Lane 1, 1 kb DNA ladder; lane 2, plasmid pSupFG1 (P) used as a control; and lane 3, TFO-directed ICL-containing pSupFG1 (P-ICL). The plasmids harboring ICLs migrate more slowly in the gel as demonstrated in lane 3 (indicated by the black arrow on the right hand side of the gel). (B) Densitometric quantification of the bands in lane 2 and lane 3 indicate that approximately 70% of the plasmids are crosslinked. This figure has been modified from reference16. Please click here to view a larger version of this figure.
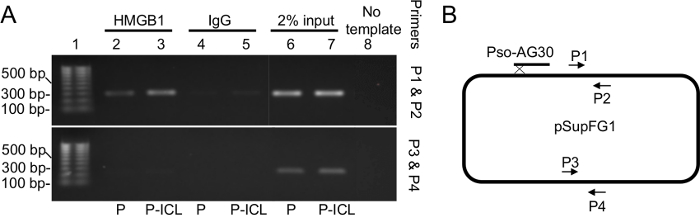
Figure 2: ChIP assay demonstrating enrichment of HMGB1 on the TFO-directed ICL-containing region of the crosslinked plasmid. (A) PCR amplification of the immunoprecipitated fragments were resolved on a 1% agarose gel using a set of proximal primers near the TFO-directed ICL site (B; P1 and P2, top panel) and a second set of primers used as a specificity control that were further (approximately 2,000 bp) from the site-directed ICL (B; P3 and P4, bottom panel). Lanes 2 and 3 indicate enrichment of HMGB1 near the TFO-directed ICL (lane 3) relative to undamaged DNA (lane 2). Lanes 4 and 5 are antibody specificity controls using an IgG antibody. Lanes 6 and 7 are the input samples. Lane 8 is a negative control for the PCR reaction and contains no DNA template. P, plasmid pSupFG1. P-ICL, TFO-directed ICL-containing plasmid pSupFG1. This figure has been modified from reference 16. (B) A schematic diagram of the plasmid pSupFG1 showing the primer sites for P1 and P2 as well as P3 and P4. Please click here to view a larger version of this figure.
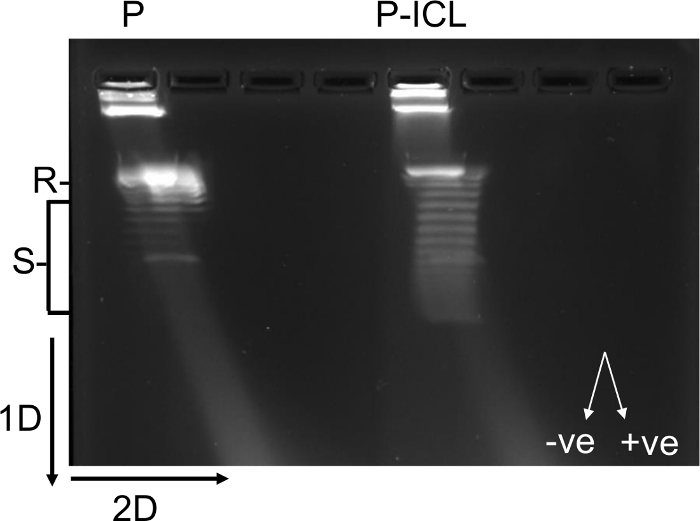
Figure 3: 2D agarose gel electrophoresis showing formation of negative supercoils in TFO-directed ICL-containing plasmids, facilitated by HMGB1. The 1x TBE agarose gel reveals the thermal distribution of the topoisomers generated by the plasmid alone (P) or the psoralen-crosslinked plasmid (P-ICL). The mobility of the supercoiled species is faster compared to the relaxed plasmids. Each band represents a topoisomer that differs from the band below or above by one less or more superhelical turn, respectively. The left-handed arc represents negatively supercoiled (-ve) species and the right-handed arc represents positively supercoiled (+ve) species. The psoralen ICL-containing plasmid population (P-ICL) contains more supercoiled species than the control plasmids (P). R, relaxed plasmids; S, supercoiled plasmids; 1D, first dimension of the gel electrophoresis; 2D, second dimension of the gel electrophoresis. This figure has been modified from16. Please click here to view a larger version of this figure.
