GST-BMI1 and RING1B proteins are well produced in bacteria and can be readily extracted in the soluble fraction. Figure 1A shows a Coomassie blue staining for a typical purification of the GST-BMI1-RING1B complex. The GST-BMI1 and RING1B bands migrate at the expected molecular weight, ~45 kDa and ~13 kDa respectively. Notably the E3 ligase complex is highly homogenous with very low levels of bacteria proteins contaminants and/or degradation products. Moreover, the stoichiometry of the purified complex is optimal, as with a molar ratio of 1:1, the band intensity of GST-RING1B protein is expected to be two to three times stronger than that of BMI1. Similarly, the purification of His-BAP1/MBP-DEUBAD complex also resulted in relatively highly homogenous preparations with bands at ~90 kDa and ~55 kDa, respectively (Figure 1B). The tandem affinity purification of BAP1 and DEUBAD, through nickel-agarose and amylose-agarose respectively ensures and adequate stoichiometry and the removal of free BAP1 from the purified complex. On the other hand, the purified nucleosomal fraction is essentially composed by 147bp band indicating the presence of highly enriched mononucleosomal fraction (Figure 1C). Coomassie blue staining of these purified nucleosomes shows a typical band pattern of the four histone subunits with stoichiometric amounts (Figure 1D). Commercially available recombinant mononucleosomes also display similar protein and DNA patterns when migrated on SDS-PAGE and agarose gels respectively. Of note, monoubiquitinated forms of H2A and Flag-H2A can be readily distinguished on HEK293T-purified nucleosomes. We used recombinant nucleosomes along with E1/E2/Ubiquitin/ATP and observed that ubiquitination of histone H2A with the BMI1-RING1B complex shows a time-dependent increase of the ubiquitinated form of the protein, while the levels of the non-modified form are concomitantly decreased. The ubiquitination reaction is total, as virtually all H2A proteins are transformed to H2A K119ub (Figure 1E). On the other hand, deubiquitination assay was conducted on native nucleosomes. Following incubation of these nucleosomes with BAP1/DEUBAD, we observed a time-dependent-reduction of H2A K119ub signal (Figure 1F). Note that two bands of ubiquitinated H2A observed with anti-H2A K119ub correspond to transfected Flag-H2A and endogenous H2A migrating at ~30 kDa and ~25 kDa bands respectively.
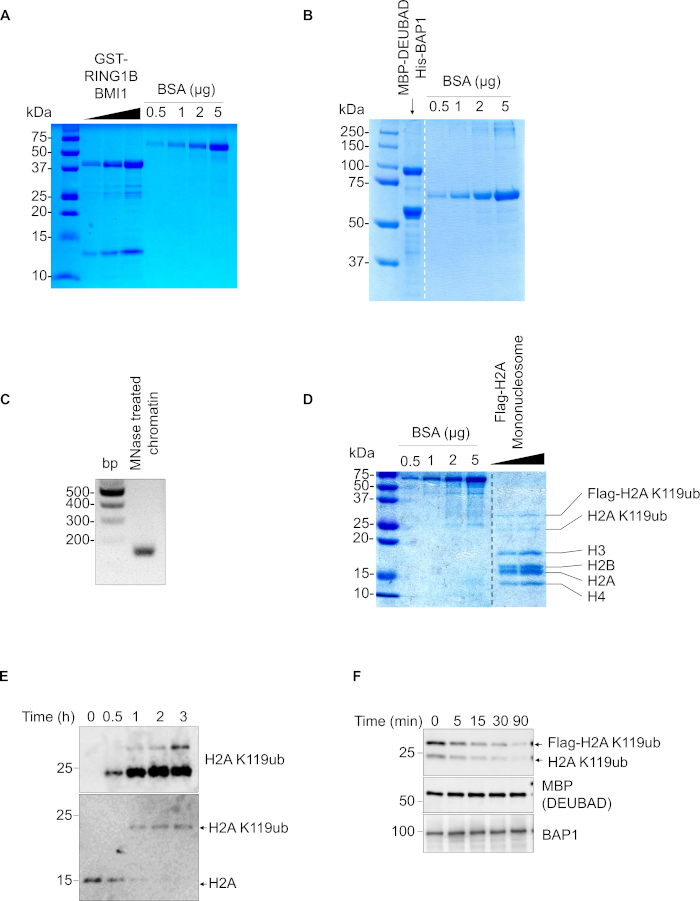
Figure 1: Purification, ubiquitination, and deubiquitination. (A) Purification of GST-RING1B-BMI1 proteins and analysis using Coomassie blue staining. GST-RING1B-BMI1 were produced in bacteria and purified using GST affinity beads. To confirm purification purity and proteins size, elutions were loaded on 15% SDS-PAGE gel and stained with Coomassie blue. Different amount of BSA are used to determine protein quantity. The stained gel was scanned to generate the figure. (B) Purification of MBP-DEUBAD/His-BAP1 complex. MBP-DEUBAD and HIS-BAP1 were produced separately in bacteria. After lysis, MBP-DEUBAD and HIS-BAP1 were mixed and purified using nickel and maltose beads. The purified complex was loaded on 8% SDS-PAGE gel and stained with Coomassie blue. (C) Purification of mononucleosome following MNase treatment. Chromatin from HEK293T expressing pcDNA.3 Flag-H2A was extracted and treated with MNase to generate and purify mononucleosomes. To confirm the generation of mononucleosome, a fraction of chromatin was taken for phenol chloroform extraction and detection under UV light. Loading on 2% agarose gel reveals a typical 147 base-pair DNA fragment indicative of mononucleosome. (D) Purified mononucleosomes were used for Coomassie blue staining. (E) In vitro ubiquitination of nucleosomes. Recombinant nucleosomes were incubated at 37 °C with the bacterial purified E3 ubiquitin ligase dimer, GST-RING1B/BMI1 and E1/E2/ATP/Ubiquitin, for the indicated times. H2A monoubiquitination (H2A K119ub) was analysed by western blotting. (F) Deubiquitination assay of H2A K119ub using the BAP1/DEUBAD deubiquitinase complex. In vitro deubiquitination assays were conducted using bacteria purified His-BAP1/MBP-DEUBAD and mammalian purified nucleosomes. Reactions were done for the indicted times and analyzed by western blotting. Please click here to view a larger version of this figure.
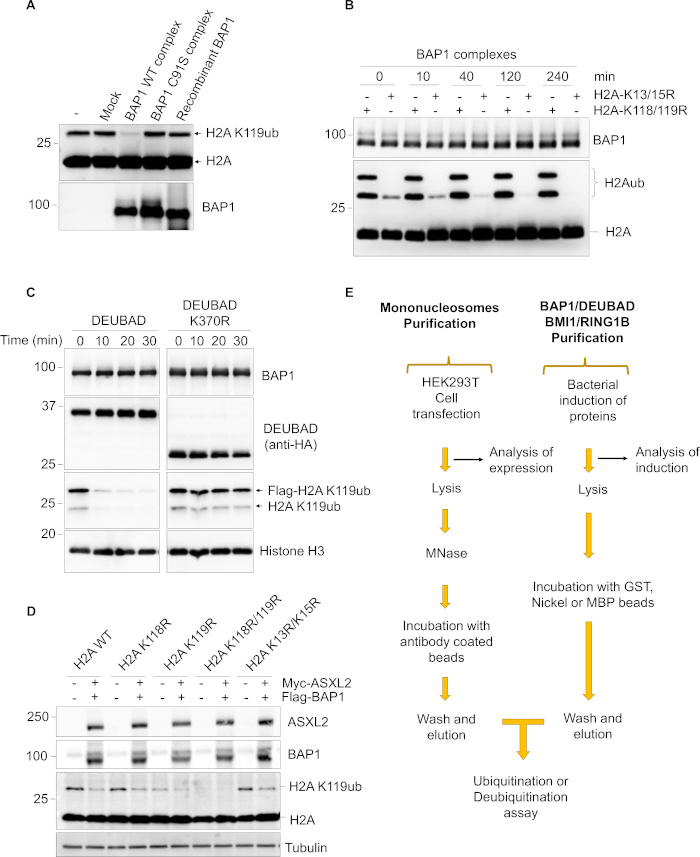
Figure 2: Controls and overview of the protocol. (A) Purified mammalian BAP1 or its catalytic dead mutant (C91S) as well as bacteria-purified recombinant BAP1 were used for H2A deubiquitination on nucleosomes. Nucleosomes incubated with buffer alone or with elutions obtained from the mock purification were also included as controls. (B) BAP1 does not deubiquitinate H2A K13/K15 whose ubiquitination is mediated by RNF168 E3 ligase. HEK293T cells were co-transfected with either H2A K13R/K15R or H2A K118R/K119R along with RNF168 to ensure ubiquitination on K13/K15 residues. The purified nucleosomes were used for deubiquitination by the BAP1 complexes. The reaction was stopped at different time points and subjected to immunoblotting. (C) Purification of non-ubiquitinated and monoubiquitinated DEUBAD in complex with BAP1 from mammalian cells. The purified complex was used for deubiquitination assay on nucleosomal H2A K119ub. Reactions were done for the indicated time points and analyzed by western blotting. (D) BAP1 deubiquitinates H2A K119ub in vivo. HEK293T cells were co-transfected with H2A or H2A mutants (H2A K118R, K119R, K118R/K119R or K13R/K15R) along with BAP1 and ASXL2 constructs. Two days post transfection, cells were harvested for immunoblotting using the indicated antibodies. (E) Schematic representation of the experimental workflow. Please click here to view a larger version of this figure.
