Het begrijpen van representaties van de moleculaire wereld is van cruciaal belang om een expert te worden in de biomoleculairewetenschappen 1, omdat de interpretatie van dergelijke beelden de sleutel is tot het begrijpen van de biologische functie2. De inleiding van een leerling tot macromoleculen komt meestal in de vorm van tweedimensionale leerboekafbeeldingen van celmembranen, organellen, macromoleculen, enz., Maar de biologische realiteit is dat dit driedimensionale structuren zijn, en een goed begrip van hun eigenschappen vereist manieren om betekenis te visualiseren en te extraheren uit 3D-modellen.
Dienovereenkomstig heeft de ontwikkeling van biomoleculaire visuele geletterdheid in hogere divisie moleculaire life science-cursussen aandacht gekregen, met een aantal artikelen die rapporteren over het belang en de moeilijkheden van het onderwijzen en beoordelen van visualisatievaardigheden1,3,4,5,6,7,8,9 . De reactie op deze artikelen is een toename van het aantal klassikale interventies, meestal binnen een semester in een enkele instelling, waarbij moleculaire visualisatieprogramma’s en -modellen worden gebruikt om moeilijke concepten te targeten2,10,11,12,13,14,15 . Daarnaast hebben onderzoekers geprobeerd te karakteriseren hoe studenten biomoleculaire visualisatieprogramma’s en / of modellen gebruiken om een specifiek onderwerp16,17,18,19te benaderen. Onze eigen groep, BioMolViz, heeft een raamwerk beschreven dat overkoepelende thema’s in visuele geletterdheid onderverdeelt in leerdoelen en doelstellingen om dergelijke interventies te begeleiden20,21,en we leiden workshops die faculteiten trainen om het kader te gebruiken in het achterwaartse ontwerp van beoordelingen om visuele geletterdheidsvaardigheden te meten22.
Centraal in al dit werk staat een kritische vaardigheid: het vermogen om structuren van macromoleculen te manipuleren met behulp van programma’s voor biomoleculaire visualisatie. Deze tools zijn onafhankelijk ontwikkeld met behulp van verschillende platforms; daarom kunnen ze vrij uniek zijn in hun werking en gebruik. Dit vereist programmaspecifieke instructies en de identificatie van een programma waarmee een gebruiker vertrouwd is, is belangrijk om de verdere implementatie te vergemakkelijken.
Naast de basisprincipes van het manipuleren van structuren in 3D (roteren, selecteren en wijzigen van het model), is een belangrijk doel om de actieve plaats van een eiwit te modelleren. Dit proces stelt een leerling in staat om zijn begrip te ontwikkelen in drie overkoepelende thema’s beschreven door het BioMolViz Framework: moleculaire interacties, liganden / modificaties en structuur-functierelaties20,21.
Vier populaire keuzes van programma’s voor biomoleculaire visualisatie zijn: Jmol / JSmol23,iCn3D24,PyMOL25en UCSF Chimera26,27. We moedigen degenen die nieuw zijn bij Chimera aan om UCSF ChimeraX te gebruiken, de volgende generatie van het Chimera moleculaire visualisatieprogramma, de momenteel ondersteunde versie van het programma.
In dit protocol laten we zien hoe we elk van deze vier programma’s kunnen gebruiken om de actieve plaats van menselijk glucokinase te modelleren met een gebonden substraat analoog complex (PDB ID: 3FGU), en om metingen weer te geven om specifieke bindingsinteracties te illustreren28. Het model vertegenwoordigt een katalytisch complex van het enzym. Om de actieve plaats in de prekatalysetoestand vast te leggen, werd een niet-hydrolyseerbaar analoog van ATP gebonden aan de actieve glucokinase-plaats. Deze fosfoaminofosfonzuur-adenylaatester (ANP) bevat een fosfor-stikstofbinding in plaats van de gebruikelijke fosfor-zuurstofkoppeling op deze positie. De actieve site bevat ook glucose (aangeduid met BCG in het model) en magnesium (aangeduid met MG). Bovendien is er een kaliumion (K) in de structuur, als gevolg van kaliumchloride dat wordt gebruikt in het kristallisatie-oplosmiddel. Dit ion is niet kritisch voor de biologische functie en bevindt zich buiten de actieve plaats.
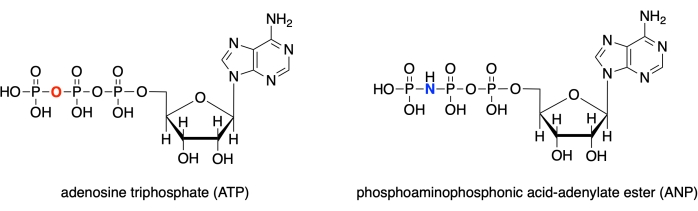
Figuur 1: ATP/ANP structuren. Adenosinetrifosfaat (ATP) structuur vergeleken met de fosfoaminofosfonzuur-adenylaat ester (ANP). Klik hier om een grotere versie van deze figuur te bekijken.
Het protocol toont de selectie van de gebonden liganden van het substraat analoog complex en de identificatie van actieve-site residuen binnen 5 Å van het gebonden complex, dat aminozuren en watermoleculen vangt die in staat zijn om relevante moleculaire interacties te maken, waaronder hydrofobe en van der Waals interacties.
De weergave wordt in eerste instantie gemanipuleerd om het grootste deel van het eiwit in een cartoonweergave weer te geven, met de aminozuurresiduen van de actieve plaats in stokweergave om de relevante atomen van het eiwit te tonen en de moleculaire interacties te benadrukken. Na stap 3 van het protocol voor elk programma zijn deze representaties toegepast en is de weergave van het eiwit vergelijkbaar tussen programma’s(figuur 2). Aan het einde van het protocol is de eiwitcartoon verborgen om de weergave te vereenvoudigen en zich te concentreren op de actieve site.
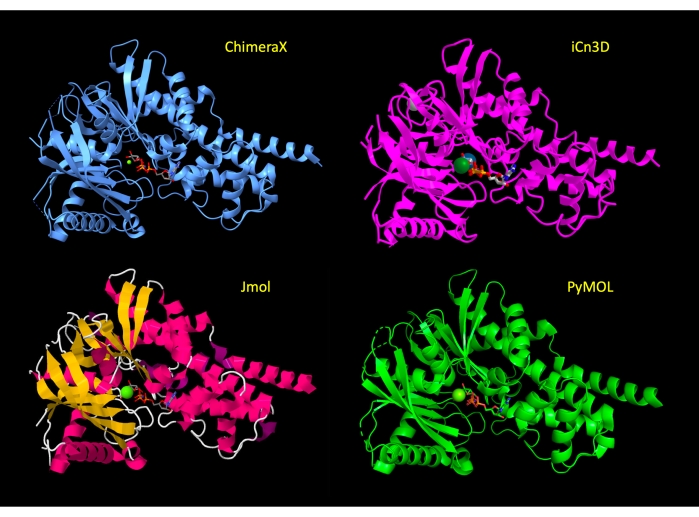
Figuur 2: Structuurvergelijking tussen programma’s. Vergelijking van de structuur van 3FGU in elk programma volgens de stap De representatie aanpassen (stap 2 of 3 van elk protocol). Klik hier om een grotere versie van deze figuur te bekijken.
CPK-kleuring wordt toegepast op de actieve plaats aminozuren en gebonden liganden29,30. Dit kleurschema onderscheidt atomen van verschillende chemische elementen in moleculaire modellen die worden weergegeven in lijn-, stok-, bal- en stok- en ruimtevullende representaties. Waterstof is wit, stikstof is blauw, zuurstof is rood, zwavel is geel en fosfor is oranje in het CPK-kleurenschema. Traditioneel wordt zwart gebruikt voor koolstof, hoewel in modern gebruik de koolstofkleuring kan variëren.
Waterstofatomen zijn niet zichtbaar in kristalstructuren, hoewel elk van deze programma’s in staat is om hun locatie te voorspellen. Het toevoegen van de waterstofatomen aan een grote macromoleculaire structuur kan het zicht vertroebelen, dus ze worden niet weergegeven in dit protocol. Dienovereenkomstig zullen waterstofbruggen worden getoond door te meten vanuit het midden van twee heteroatomen (bijv. Zuurstof naar zuurstof, zuurstof naar stikstof) in deze structuren.
Programma Overzichten
Downloadbare grafische gebruikersinterfaces (GUI’s): PyMOL (versie 2.4.1), ChimeraX (versie 1.2.5) en Jmol (versie 1.8.0_301) zijn gui-gebaseerde moleculaire modelleringstools. Deze drie interfaces zijn voorzien van opdrachtregels om getypte code in te voeren; veel van dezelfde mogelijkheden zijn beschikbaar via menu’s en knoppen in de GUI. Een gemeenschappelijk kenmerk in de opdrachtregel van deze programma’s is dat de gebruiker eerdere opdrachten kan laden en opnieuw kan uitvoeren met behulp van de pijltoetsen omhoog en omlaag op het toetsenbord.
Webgebaseerde GUI’s: iCn3D (I-see-in-3D) is een WebGL-gebaseerde viewer voor interactieve weergave van driedimensionale macromoleculaire structuren en chemicaliën op het web, zonder dat u een afzonderlijke toepassing hoeft te installeren. Het maakt geen gebruik van een opdrachtregel, hoewel de volledige webversie een bewerkbaar opdrachtlogboek bevat. JSmol is een JavaScript- of HTML5-versie van Jmol voor gebruik op een website of in een webbrowservenster en lijkt qua werking sterk op Jmol. JSmol kan worden gebruikt om online tutorials te maken, inclusief animaties.
Proteopedia31,32, FirstGlance in Jmol33en de JSmol-webinterface (JUDE) aan de Milwaukee School of Engineering Center for BioMolecular Modeling zijn voorbeelden van dergelijke Jmol-gebaseerde online ontwerpomgevingen34. De Proteopedia wiki is een leermiddel waarmee de gebruiker een macromolecuulstructuur kan modelleren en pagina’s met deze modellen kan maken binnen de website35. De Proteopedia scene authoring tool, gebouwd met behulp van JSmol, integreert een GUI met extra functies die niet beschikbaar zijn in de Jmol GUI.
Jmol en iCn3D zijn gebaseerd op de programmeertaal Java; JSmol gebruikt Java of HTML5 en PyMOL en ChimeraX zijn gebaseerd op de programmeertaal Python. Elk van deze programma’s laadt eiwitgegevensbankbestanden, die kunnen worden gedownload van de RCSB Protein Data Bank onder een 4-cijferige alfanumerieke PDB ID36,37. De meest voorkomende bestandstypen zijn Protein Data Bank (PDB)-bestanden met de extensie .pdb en Crystallographic Information File (CIF of mmCIF) met de extensie .cif. CIF heeft PDB vervangen als het standaardbestandstype voor de Protein Data Bank, maar beide bestandsindelingen werken in deze programma’s. Er kunnen kleine verschillen zijn in de manier waarop de volgorde/structuur wordt weergegeven bij het gebruik van CIF in tegenstelling tot PDB-bestanden; de bestanden werken echter op dezelfde manier en de verschillen zullen hier niet in detail worden behandeld. De Molecular Modeling Database (MMDB), een product van het National Center for Biotechnology Information (NCBI), is een subset van VOB-structuren waaraan categorische informatie is gekoppeld (bijv. Biologische kenmerken, geconserveerde eiwitdomeinen)38. iCn3D, een product van de NCBI, is in staat om PDB-bestanden met de MMDB-gegevens te laden.
Om een model te bekijken, kan de gebruiker het gewenste bestand downloaden van de speciale pagina Eiwitgegevensbank voor de structuur (bijvoorbeeld https://www.rcsb.org/structure/3FGU) en vervolgens het vervolgkeuzemenu Bestand van het programma gebruiken om de structuur te openen. Alle programma’s zijn ook in staat om een structuurbestand rechtstreeks via de interface te laden, en die methode wordt gedetailleerd in de protocollen.
De ChimeraX-, Jmol- en PyMOL-GUI’s bevatten elk een of meer vensters van de console die kunnen worden gewijzigd door de hoek te slepen. iCn3D en JSmol bevinden zich volledig in een webbrowser. Wanneer u iCn3D gebruikt, moet de gebruiker mogelijk in de pop-upvensters scrollen om alle menu-items weer te geven, afhankelijk van de schermgrootte en resolutie.
De protocollen die hier worden beschreven, bieden een eenvoudige methode om de actieve plaats van het enzym weer te geven met behulp van elk programma. Opgemerkt moet worden dat er meerdere manieren zijn om de stappen in elk programma uit te voeren. In ChimeraX kan dezelfde taak bijvoorbeeld worden uitgevoerd met behulp van vervolgkeuzemenu’s, de werkbalk bovenaan of de opdrachtregel. Gebruikers die geïnteresseerd zijn in het leren van een specifiek programma in detail worden aangemoedigd om de online tutorials, handleidingen en Wiki’s te verkennen die beschikbaar zijn voor deze programma’s39,40,41,42,43,44,45,46.
Bestaande handleidingen en zelfstudies voor deze programma’s presenteren de items in dit protocol als afzonderlijke taken. Om een actieve site weer te geven, moet de gebruiker de vereiste bewerkingen synthetiseren uit de verschillende handleidingen en zelfstudies. Dit manuscript vormt een aanvulling op bestaande zelfstudies die beschikbaar zijn door een lineair protocol te presenteren voor het modelleren van een gelabelde actieve site met moleculaire interacties, waardoor de gebruiker een logica krijgt voor actieve sitemodellering die kan worden toegepast op andere modellen en programma’s.
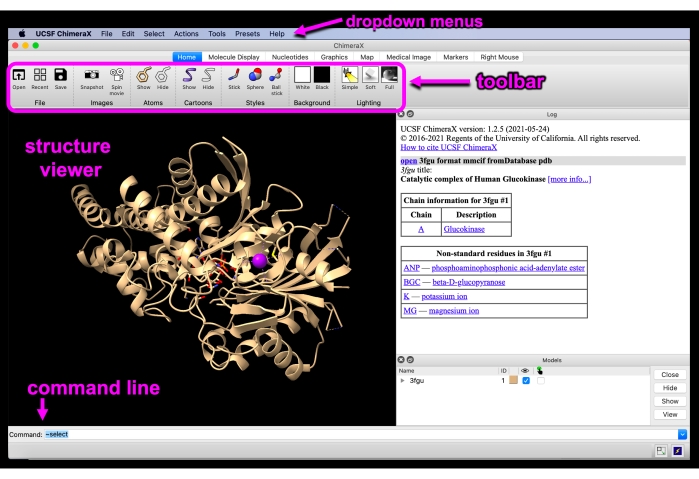
Figuur 3: Chimerax GUI. ChimeraX GUI-interface met de vervolgkeuzemenu’s, werkbalk, structuurviewer en opdrachtregel gelabeld. Klik hier om een grotere versie van deze figuur te bekijken.
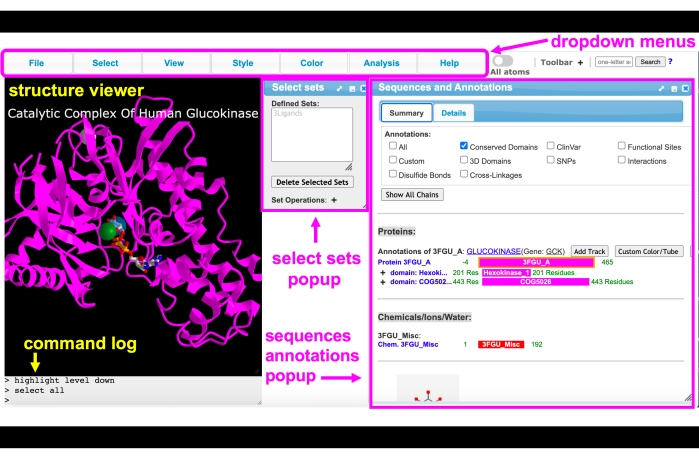
Afbeelding 4: iCn3D GUI. iCn3D GUI-interface met de vervolgkeuzemenu’s, werkbalk, structuurviewer, opdrachtlogboek, pop-up van geselecteerde sets en pop-upmenu’s met volgorde en annotaties gelabeld. Klik hier om een grotere versie van deze figuur te bekijken.
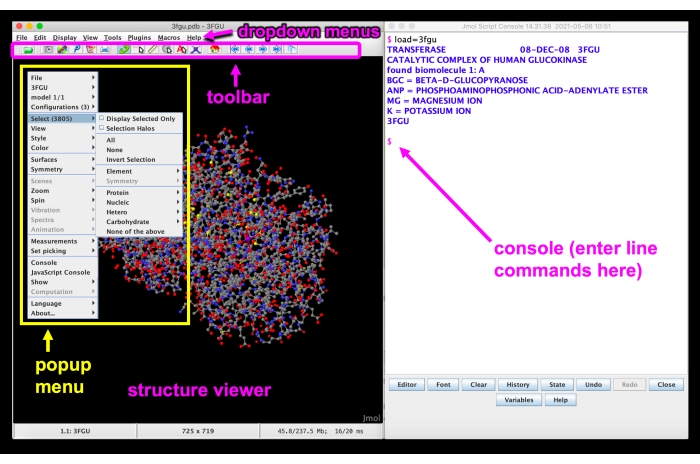
Figuur 5: Jmol GUI. Jmol GUI-interface met de vervolgkeuzemenu’s, werkbalk, structuurviewer, pop-upmenu en console / opdrachtregel gelabeld. Klik hier om een grotere versie van deze figuur te bekijken.

Figuur 6: PyMOL GUI. PyMOL GUI-interface met de vervolgkeuzemenu’s, structuurviewer, namen / objectpaneel, muisbedieningsmenu en opdrachtregel gelabeld. Klik hier om een grotere versie van deze figuur te bekijken.
