1. µSiM device assembly
NOTE: This method describes the assembly of the devices. The membrane chip has a trench-side and flat-side, which can be assembled trench-up or trench-down. Trench-down devices are most commonly used for cell culture.
- In a sterile environment (i.e., a biosafety hood), prepare all the materials for assembly, including the assembly fixtures, components, membrane chips, and tweezers.
- Place the membrane chip onto the center of fixture A1 using chip tweezers. The flat surface of the chip faces down and the trench area faces up for trench-down devices, and vice versa for trench-up devices.
NOTE: The following assembly steps are illustrated by using the trench-down device as an example, which allows a flat growth area in the top chamber. - Bond component 1 to the chip. First, peel off the blue protective layers on both sides of component 1 using straight tweezers and place it in fixture A1, top chamber down. Gently press component 1 down until the pressure-sensitive adhesive (PSA) touches the chip.
- Put fixture A2 onto fixture A1 and apply firm pressure at different corners to ensure a tight fit of the chip and component.
NOTE: Be sure to not take the PSA layer off. It is a transparent and stiffer layer compared to the blue protective layers. - Bond component 2 to component 1 with the chip. First, prepare component 2 by grabbing one corner of component 2 with straight tweezers and peeling it off from the sheet. Then, grab the non-PSA "triangle" region of component 2 and together remove the thick, transparent protective layer and blue layer of component 2, exposing the PSA surface. Place component 2 in fixture B1, PSA side up.
- Place component 1 with the membrane chip in fixture B1, top chamber facing up.
- Put fixture B2 onto component 1 and apply firm pressure at different corners of fixture B2.
- Take the assembled device out of the fixture and use straight tweezers to press out any air bubbles on the underside of the device and seal the edges of the channelx`, avoiding contact with the membrane region. Prior to using for cell culture, ultraviolet (UV)-sterilize newly assembled devices for 20 min.
2. Cell culture
NOTE: This method describes protocols for primary and hiPSC-derived cultures on the platform. The methods describe endothelial cell monoculture in the top chamber of the device and pericyte and endothelial cell co-culture with pericytes in the bottom chamber and endothelial cells in the top chamber of trench-down assembled devices. For chamber dimensions and volumes, see Table 1. These are the most common formats; however, other cell culture layouts can be used depending on user needs.
- Cell culture chamber preparation
- Assemble the devices per the protocol detailed in section 1.
- Place the devices in sterile hose clamps. Prior to cell culture, sterilize the clamps by soaking them in ethanol for ≥20 min. Resterilize after each experiment for reuse.
- Culture the devices in a large sterile Petri dish. For extra humidity, add a small Petri dish or 50 mL conical tube lid filled with sterile water.
- Wash the top chamber of the devices twice with 100 µL of sterile water. For bottom chamber cell culture, wash the bottom chamber with 20 µL of sterile water.
- hCMEC/D3 cell line (BBB) monoculture
- Prepare the cell culture chamber according to section 2.1.
- Coat the top chambers with 25 µg/cm2 of collagen I and 5 µg/cm2 of human fibronectin mixed in phosphate-buffered saline (PBS). Let it sit for 1-2 h at 37 °C or overnight at 4 °C.
- Remove the coating solution and add 100 µL of prewarmed growth-factor-depleted endothelial medium (Assay medium) to the top chamber and 20 µL to the lower chamber. To prepare the assay medium, mix 100 mL of endothelial basal medium + 400 µL of human fibroblastic growth factor + 40 µL of hydrocortisone + 100 µL of gentamicin sulfate + 2 mL of fetal bovine serum.
- Passage hCMEC/D3 cells according to the manufacturer's protocol; trypsinize the cells in a 37 °C incubator for 3-5 min and then stop trypsinization by the addition of prewarmed endothelial medium. Transfer the cell suspension to a centrifuge tube and centrifuge at 200 × g for 5 min at room temperature. Resuspend the cell pellet in assay media and seed the cells in the top chambers at a density of 40,000-60,000 cells/cm2. Incubate the devices at 37 °C with 5% carbon dioxide (CO2) for 2-4 h to facilitate cell adhesion. For example, to achieve 40,000 cells/cm2 on the 0.37 cm2 chip, add 100 µL of a cell solution with a concentration of 150,000 cells/mL to the top chamber.
NOTE: We culture and passage hCMEC/D3 according to Hudecz et al.38, using a modified formulation of endothelial growth media-2 (EGM-2) for cell maintenance and a growth factor-reduced "assay medium" after subculture for assays. Other media formulations should be compatible with the devices, though we recommend the media formulations from Hudecz et al.38 if users experience issues with hCMEC/D3 survival. - After 2-4 h for cell adhesion, exchange with fresh assay medium in both chambers to remove dead or unattached cells.
- Maintain the devices at 37 °C with 5% CO2, exchanging the assay medium in both chambers every 2-3 days until experimentation. Assays are typically performed after 2 weeks of culture.
- hiPSC-derived EECM-BMEC-like cell monoculture
- Prepare the cell culture chamber according to section 2.1.
- Prepare collagen IV from human placenta solution by adding 5 mL of 0.5 mg/mL acetic acid to 5 mg of collagen IV to create a 1 mg/mL solution. Let the solution sit for ≥4 h at 4 °C to fully reconstitute. The solution is stable at 4 °C for 2 weeks.
- Coat the top chambers with 100 µL of a 4:1:5 ratio of collagen IV, bovine fibronectin, and sterile water. Coat at 37 °C for 2-4 h.
- Remove the coating solution and add 100 µL of room temperature hECSR to the top chamber and 20 µL to the lower chamber.
- Passage EECM-BMEC-like cells according to Nishihara et al.37; add a cell detachment enzyme mixture to the cells and transfer to a 37 °C incubator for 5-8 min. Pipet the cell solution to singularize and add to 4x the volume of endothelial medium in a centrifuge tube. Centrifuge at 200 × g for 5 min at room temperature; resuspend the cells in hECSR and seed in the top chambers at a density of 40,000 cells/cm2. Incubate the devices at 37 °C with 5% CO2 for 2-4 h to facilitate cell adhesion. For example, to achieve 40,000 cells/cm2, add 100 µL of a cell solution at 150,000 cells/mL.
- After 2-4 h for cell adhesion, exchange with fresh hECSR in both chambers to remove dead or unattached cells.
- Maintain the devices at 37 °C with 5% CO2, exchanging hECSR in both chambers every 1-2 days until experimentation. Assays are typically performed on day 6 of culture.
- hCMEC/D3 and primary human brain vascular pericyte (HBVP) co-culture
- Prior to the device assembly, coat the membrane chips with 2 µg/cm2 of poly-L-lysine (PLL) mixed in PBS. Apply 50-80 μL PLL in a droplet only to the side that will be facing down in the final device assembly. Complete the coating process in either 1-2 h at 37 °C or overnight at 4 °C.
- Remove the coating solution. Wash the membrane chips with sterile ultrapure water and allow them to dry.
- Assemble the devices per the protocol detailed in section 1 above, with the PLL-coated side facing down, and prepare the cell culture chamber according to section 2.1.
- Coat the top chambers according to section 2.2.2.
- Add 50 µL of prewarmed pericyte medium to the top chambers and add 20 µL to the lower channels.
- Passage HBVPs according to the manufacturer's protocol; trypsinize the cells in a 37 °C incubator for 3-5 min, then stop trypsinization by addition of prewarmed pericyte medium. Transfer the cell suspension to a centrifuge tube and centrifuge at 200 × g for 5 min at room temperature. Resuspend the cell pellet in pericyte medium and seed the cells in the bottom chambers at a density of 14,000-25,000 cells/cm2. Invert the devices but maintain an air interface with the top chambers to facilitate gas exchange.
NOTE: The air interface required during inversion can be achieved by flipping the devices after placing them inside hose clamps and pushing down on all corners prior to flipping, or by placing the devices upside down on parallel strips of acrylic or silicone spaced far enough apart to keep the top chambers unobstructed. Seeding density may need to be optimized for each user. To achieve 14,000 cells/cm2, add 20 µL of a cell suspension at ~590,000 cells/mL. Note that 20 µL is pipetted into the bottom chamber to avoid bubbles, but the density is calculated using the bottom chamber volume of 10 µL. - Incubate the cells at 37 °C with 5% CO2 for 2-4 h in the inverted position to facilitate cell adhesion.
- After 2-4 h for cell adhesion, flip the devices upright and exchange them with fresh pericyte medium in both chambers to remove dead or unattached cells.
- After pericyte seeding, seed hCMEC/D3s in the top chamber, following steps 2.2.4-2.2.5. Switch both chambers to the assay medium.
- Maintain the devices at 37 °C with 5% CO2, exchanging assay medium in both chambers every 1-2 days until experimentation. Primary cell co-culture is usually maintained for 6-8 days prior to the assays.
NOTE: If the HBVP monocultures will be compared to hCMEC/D3 monocultures or hCMEC/D3 and HBVP co-cultures, then exchange pericyte medium with assay medium 2-3 days after seeding the HBVPs.
- hiPSC-derived EECM-BMEC-like cell and brain pericyte-like cell (BPLC) co-culture
- Prepare the cell culture chamber according to section 2.1 and coat the top chambers according to steps 2.3.2-2.3.3.
- Remove the coating solution and add 50 µL of room temperature Essential 6 Medium + 10% Fetal Bovine Serum (E6 + 10% FBS) to the top chamber.
- Passage the BPLCs according to Gastfriend et al.39; add a cell detachment enzyme mixture to the cells and transfer to a 37 °C incubator for 5-15 min, until ~90% of the cells are rounded. Pipet the cell solution to singularize and add 4x the volume of DMEM/F12 in a 50 mL centrifuge tube, using a 40 µm cell strainer to filter and fully singularize the cells. Transfer to a 15 mL centrifuge tube, centrifuge at 200 × g for 5 min at room temperature, resuspend the cell pellet in E6 + 10% FBS, and seed the cells in the bottom chambers at a density of 14,000-25,000 cells/cm2. Invert the devices but maintain an air interface with the top chamber to facilitate gas exchange.
NOTE: The air interface required during inversion can be achieved by flipping the devices after placing them inside the hose clamps and pushing down on all corners prior to flipping, or by placing the devices upside down on parallel strips of acrylic or silicone spaced far enough apart to keep the top chambers unobstructed. Seeding density may need to be optimized for each user. To achieve 14,000 cells/cm2, add 20 µL of a cell suspension at ~590,000 cells/mL. Note that 20 µL is pipetted into the bottom chamber to avoid bubbles, but the density is calculated using the bottom chamber volume of 10 µL. - Incubate the cells at 37 °C with 5% CO2 for 2-4 h in the inverted position to facilitate cell adhesion.
- After 2-4 h for cell adhesion, flip the devices upright and exchange with fresh E6 + 10% FBS in both chambers to remove dead or unattached cells.
- One day after pericyte seeding, seed EECM-BMEC-like cells in the top chamber, following steps 2.3.5-2.3.6. Switch both chambers to hECSR.
- Maintain the devices at 37 °C with 5% CO2, exchanging the assay medium in both chambers every 1-2 days until experimentation. hiPSC-derived co-culture is usually maintained for 7 days for BPLCs and 6 days for EECM-BMEC-like cells prior to assays.
NOTE: If the BPLC monocultures will be compared to EECM-BMEC monocultures or EECM-BMEC and BPLC co-cultures, then exchange E6 + 10% FBS with hECSR 1 day after seeding the BPLCs.
3. Immunocytochemistry
NOTE: This method describes a protocol for immunocytochemical staining and imaging of cells cultured in the top and/or bottom side of the membrane. The aim of this experiment is to determine the presence and location of key proteins that should be found in the BBB such as adherens and tight junction proteins, and cell identity proteins. Alternate and live staining methods are also compatible with the platform.
- Fixation and staining on the devices
- Place the primary antibodies on ice to thaw.
- Prepare a 4% paraformaldehyde (PFA) solution at room temperature (e.g., dilute 16% PFA in 3x its volume of PBS) or chill 100% methanol at -20 °C.
- Create an appropriate blocking solution according to Table 2. Store on ice.
NOTE: Vortexing the solution may be necessary to fully dissolve Triton X-100. - Fix the cells by pipetting 20 µL of fixative (e.g., PFA or methanol) into the bottom chamber and 50 µL into the top chamber. Incubate the devices for 10 min (PFA) or 2 min (methanol) at room temperature.
- Wash for 3 x 5 min by pipetting 20 µL of PBS through the bottom chamber and 100 µL into the top chamber.
- Block for 30 min at room temperature by adding 20 µL of the blocking solution to the bottom chamber and 50 µL of blocking solution into the top chamber. Check for bubbles in the bottom chamber.
- Prepare the primary antibody solution by diluting the antibody(ies) in the blocking solution according to Table 2. Store on ice.
- Stain with the primary antibodies by adding 20 µL of the primary antibody solution to the bottom chamber and replacing the volume of the top chamber with 50 µL of the primary antibody solution. Check for bubbles in the bottom chamber. Incubate for 1 h at room temperature or overnight at 4 °C.
- Wash for 3 x 5 min by pipetting 20 µL of PBS through the bottom chamber and 100 µL into the top chamber.
- Prepare the secondary antibody solution by diluting the antibody(ies) in the blocking solution according to Table 2. Store on ice protected from light.
- Stain with secondary antibodies by adding 20 µL of secondary antibody solution to the bottom chamber and replacing the volume of the top chamber with 50 µL of the secondary antibody solution. Check for bubbles in the bottom chamber. Incubate for 1 h at room temperature protected from light.
- Wash for 3 x 5 min by pipetting 20 µL of PBS through the bottom chamber and 100 µL into the top chamber.
- Make a nuclear stain solution. Dilute Hoechst 1:10,000 in PBS. Add 20 µL of the nuclear stain solution to the bottom chamber and replace the volume of the top chamber with 50 µL of the nuclear stain solution. Check for bubbles in the bottom chamber. Incubate for 3 min at room temperature protected from light.
- Remove the stain by adding 20 µL of PBS to the bottom chamber and replacing the volume of the top chamber with 100 µL of PBS. Image the devices immediately or store them at 4 °C with the petri dish wrapped in parafilm and protected from light until ready for imaging.
- Confocal imaging
NOTE: This section describes imaging the devices using an inverted spinning disc confocal microscope with a long working distance (LWD) 40x objective (water, WD 590-610, numerical aperture 1.15) as an example. For working distances and lens compatibility, refer to supplemental material in McCloskey et al.34. Images of the entire membrane area are also taken using a 10x objective to ensure that the 40x images are representative of the entire field. These are usually taken in widefield.- Turn on the microscope and open the imaging software.
- Set the channels according to the properties of the secondary antibody and the nuclear stain, using Confocal Imaging Mode. Optimize the laser power and exposure time to ensure that the signal is above the background and reduces imaging artifacts.
- For Hoechst nuclear staining, use excitation 405, emission 450/50 Bandpass (BP), 500 ms exposure time, 50% laser power. For labels using a secondary antibody conjugated to Alexa Fluor (AF) 488, use excitation 488, emission 525/50BP, 500 ms exposure time, and 50% laser power. For labels using a secondary antibody conjugated to AF568, use excitation 561, emission 600/50BP, 500 ms exposure time, and 100% laser power.
- Place the device in the microscope slide device holder, top chamber facing up and set in the microscope using a microscope slide holder. Turn on Live and select the channel for the nuclear stain. Find the membrane using a low objective, making sure that the transparent silicon-nitride membrane region is centered, as light will not transmit through the blue, solid silicone region. Once the device is properly centered and the cell layer is found, switch to the 40x objective, wet the objective, and focus on the membrane region using the nuclear stain as a guide.
- Turn on z-stack imaging and set Scan Mode to Start/End. Set step size or count. Using the coarse adjustment knob on the microscope, set the scan start to the top of the endothelial cell layer, using the nuclear stain as a guide, and scan end at the bottom of the pericyte layer, using the nuclear stain as a guide. Check all channels to ensure that everything will be captured in the imaging field.
NOTE: We usually use auto step, which is ~0.2 µm slices. - Set the Image Name and press Acquire to start imaging. Repeat on different regions as needed.
- Process the confocal images using ImageJ40 or Imaris.
- To process in Imaris, drag the image file into the program to open. Select Section on the top menu bar to see a 2D image of the x-y plane with corresponding views of the x-z and y-z planes. Select 3D View on the top menu bar to see a 3D image. Click on the image and drag it to rotate. Select Snapshot on the top menu bar to take an image.
4. Sampling assay of small molecule permeability
NOTE: This section describes a methodology for quantitative measurements of barrier properties of the cell cultures. The aim of this experiment is to detect the concentration of a fluorescent small molecule that passes through the cell layers and enters the bottom chamber of the platform. These data are then used to calculate cellular permeability.
- Sampling technique
- Assemble the devices per the protocol detailed in section 1 and culture the desired cell lines as described in section 2. Include an additional device to serve as a cell-free, coated control device to measure the system permeability.
NOTE: Minimum three technical replicates per condition are recommended; however, only one replicate of the coated control is required. - Prior to starting the sampling assay, replace the medium at the bottom chamber with fresh medium. Check the confluency of the endothelial monolayer in each device under the microscope. Note any gaps in cell monolayers, as this will impact the diffusion of dye into the bottom chamber.
NOTE: A healthy endothelial culture should be 100% confluent. - Prepare the fluorescent small molecule solution (e.g., 150 µg/mL Lucifer Yellow, 457 Da) in cell culture medium. Prepare excess volume of the fluorescent small molecule solution to be used for the preparation of the standard solutions that serve as references to calculate the concentration of the fluorescent small molecule sampled from the bottom channel.
NOTE: We recommend the preparation of 400 µL of excess solution to be used as the standard solution with the highest concentration of the fluorescent small molecule. - Replace the medium in the top well with 100 µL of the fluorescent small molecule solution.
NOTE: We recommend using a hydrophobic pen to draw circles around the sampling ports and wait until the hydrophobic ink dries completely before the addition of fluorescent dye solution. This prevents the spread of the medium from the bottom channel around the sampling port at step 4.1.7. We recommend staggering the addition of the fluorescent small molecule solution into the top well-wait 2 min before adding the fluorescent solution to the next device, or work in groups of 3 (add the solution into three devices at the same time and wait 5 min to add the next three devices). - Incubate the devices at 37 °C, 5% CO2 for 1 h.
- During 1 h-long incubation at the previous step, prepare standard solutions by performing 2-fold serial dilutions from the fluorescent small molecule solution prepared at step 4.1.3. Pipet 50 µL of each solution into the flat-bottom black 96-well plate in triplicates. Use blank cell culture medium as the baseline standard solution.
NOTE: To prepare standard solutions, we recommend 11 serial dilutions at a final volume of 200 µL and the use of cell culture medium as blank to measure the baseline fluorescent intensity.- Transfer 200 µL of 400 µL of the excess fluorescent small molecule solution prepared at step 4.1.3 into a tube containing 200 µL of medium, mix well, and transfer 200 µL of this solution into the next tube containing 200 µL of medium. Repeat 1:2 dilutions until there are a total of 10 tubes. Pipet 50 µL of the most concentrated standard solution into column 1 in triplicates in the 96-well plate (B1, C1, D1), the 2nd most concentrated solution into column 2 (B2, C2, D2), and so on. For the 12th column in the plate, pipet 50 µL of blank media in triplicates (B12, C12, D12).
- Perform the following steps to sample the fluorescent small molecule solution from the bottom channel.
- Remove the fluorescent small molecule solution from the well to stop the process of diffusion.
- Place a tip containing 50 µL of medium into the upper port to serve as a reservoir by inserting the tip with 50 µL into the port, lifting the device, and gently ejecting the tip while holding on to the top for stabilization. Set down the device. Make sure there are no bubbles in the pipet tip or air at the tip of the pipet to avoid adding bubbles into the bottom chamber during sampling.
- Add 50 µL of medium into the top well to prevent the disruption of the cell monolayer during sampling.
- For sampling the solution from the bottom channel, push a pipetter with an empty pipet tip to its first resistance, insert the tip into the sampling port, and reverse pipet out 50 µL of the solution from the bottom channel. Transfer directly into the flat-bottom black 96-well plate containing the standard solutions.
NOTE: We recommend checking the cell monolayer under the microscope immediately after sampling. Drawing media from the bottom channel can occasionally result in disruption of the cell layer in the top well, which might cause inconsistent permeability measurements. Working quickly throughout steps in 4.1.7 will help prevent this.
- Measure the fluorescence intensity in a microplate reader using the proper excitation and emission wavelength parameters for the fluorescent small molecule used. For Lucifer Yellow, use 428 nm excitation and 536 nm emission, with optimal gain. Fluorescence is measured from the top of the plate. Add the plate into the plate reader, highlight the wells with sample (including the standard curve) and select Start to read the plate.
- Assemble the devices per the protocol detailed in section 1 and culture the desired cell lines as described in section 2. Include an additional device to serve as a cell-free, coated control device to measure the system permeability.
- Calculation of the permeability value (see Supplemental File 1 for template)
- Subtract the averaged fluorescence intensity value of the blank medium from all fluorescence intensity values measured.
- Use equation (1) to calculate the system permeability Ps:
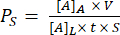 (1)
(1)
Where [A]A is the concentration of the fluorescent small molecule collected from the bottom channel, V is the volume sampled and added to the plate (0.050 mL), [A]L is the concentration of the fluorescent small molecule added into the top well (e.g. 0.15 mg/mL), t is the time of incubation (in s or min depending on the desired units), and S is the membrane surface area (0.014 cm2).- Calculate [A]A using the equation obtained by plotting a standard curve from the fluorescence intensity outputs and the known concentrations of the standard solutions. Calculate concentrations (x) of the experimental samples by inserting their fluorescence intensity values (y) into the equation.
NOTE: Use the portion of the standard curve that is linear. We recommend measuring the membrane surface area from an image of the membrane taken under the microscope as membrane area could vary depending on the lot number and may significantly affect the calculated permeability values if they differ from the coated control.
- Calculate [A]A using the equation obtained by plotting a standard curve from the fluorescence intensity outputs and the known concentrations of the standard solutions. Calculate concentrations (x) of the experimental samples by inserting their fluorescence intensity values (y) into the equation.
- Use equation (2) to calculate the permeability of the cell monolayer Pe:
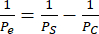 (2)
(2)
Where PS is the system permeability as calculated at step 4.2.2 and PC is the system permeability value of the cell-free, coated control device.
The assembly of a trench-down device is shown in Figure 1. The fixtures guide the assembly of the components and the membrane chip. Component 1 is primarily acrylic with a PSA surface for bonding to the chip and an opening to the bottom chamber and ports for pipet access to the bottom chamber. Component 2 is the channel layer and contains a non-adhesive, PSA-free "triangle" in the top right for gripping. Trench-down devices provide a flat cell culture growth area in the top chamber, whereas trench-up devices have a flat surface for cell culture in the bottom chamber.
We performed endothelial monocultures and co-cultures of hCMEC/D3 and HBVPs and of hiPSC IMR90-4-derived EECM-BMEC-like cells and BPLCs and acquired phase images using a Nikon Eclipse Ts2 phase contrast microscope and 10x objective (Figure 2). Optimal seeding densities for primary cell culture are shown (images taken 1 day after seeding), as well as under-seeded HBVPs (Figure 2A). Final coculture and monoculture phase images (6 days endothelial cell culture) can be difficult to distinguish (Figure 2C), and confirmation of successful primary cell co-culture may require immunofluorescence staining (see protocol section 3). Low, high, and optimal hiPSC-derived BPLC seeding densities are also illustrated (Figure 2B). hiPSC-derived co-culture is clearer to distinguish in phase contrast imaging compared to primary co-cultures (Figure 2D). Low BPLC seeding results in poor pericyte coverage and pericyte clumping, whereas overseeding results in the pericyte layer peeling off the membrane. Further, exchanging the medium in the bottom chamber too quickly may result in pericyte loss, as these cells are very sensitive to shear. Optimal coverage by pericytes is ~90% for a BBB model, with no gaps in the endothelial layer.
Representative images of immunostained hiPSC-derived co-culture are illustrated in Figure 3 (6 days endothelial cell culture). IMR90-4-derived BPLCs were stained for the pericyte marker PDGFRβ, and IMR90-4-derived EECM-BMEC-like cells were stained for the adherens junction marker VE-cadherin. Hoechst was used to stain the nuclei. Images were acquired on a Spinning Disc confocal microscope using a 40x LWD objective with 0.2 µm slices and processed with Imaris. Both cell layers can be visualized even though the thin nanomembrane is not seen.
We performed the sampling-based small molecule permeability assay using the same experimental conditions in two physically distant laboratories at the University of Bern, Switzerland, and the University of Rochester, NY, USA to demonstrate interlaboratory reproducibility of results (Figure 4)34. hiPSC IMR90-4-derived EECM-BMEC-like cells were cultured in the µSiM device for 2, 4, or 6 days and on transwell filters for 6 days. The assay was performed using 150 µg/mL Lucifer Yellow (457 Da) in both laboratories. High variability in the permeability of the endothelial cells cultured for 2 days in the device indicates that 2 days of culture was insufficient for barrier maturation. There were no significant differences in permeability between the labs upon barrier maturation-from 4 days on. We also showed that the permeability of the endothelial cells cultured in µSiM and transwell filters for 6 days matched those previously published40.
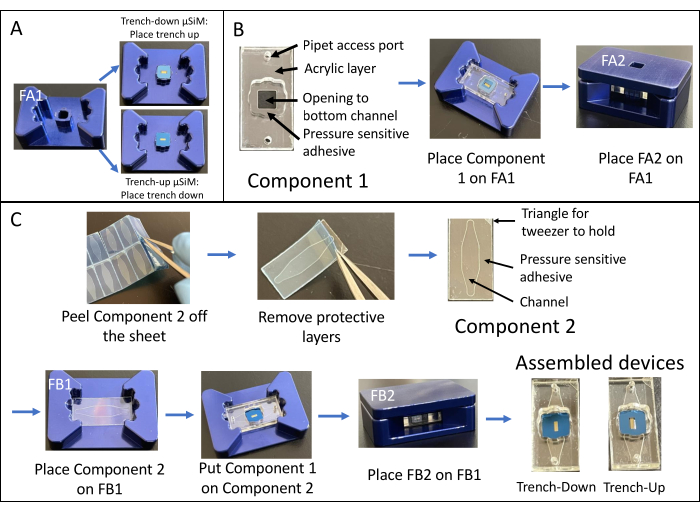
Figure 1: Steps of µSiM assembly. (A) Prepare the chip by placing it on fixture A1. Place the chip trench up for a final trench-down device. Place the chip trench down for a final trench-up device. (B) Bond component 1 to the chip by removing the protective masks from component 1 and placing it face-down into FA1. Bond by applying pressure with fixture A2. (C) Bond component 2 and component 1 by removing component 2 from its sheet and peeling off the top protective layers. Place the channel up into fixture B1 and place component 1 on top of component 2 face-up. Bond by applying pressure with fixture B2. Abbreviations: FAn = fixture An; FBn = fixture Bn. Please click here to view a larger version of this figure.
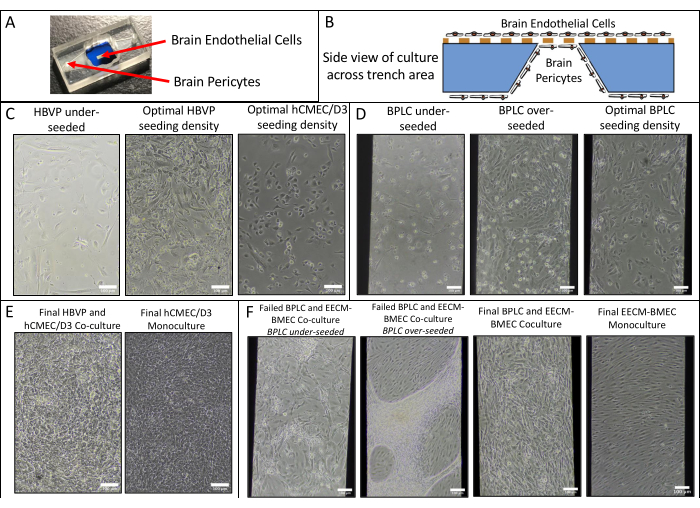
Figure 2: Schematic of coculture and representative phase contrast images of cell culture in devices. (A) Location of pericyte and endothelial cell seeding. (B) Side view schematic of cell locations across the trench of the membrane chip. (C) Representative images of low and optimal seeding densities for primary HBVP and hCMEC/D3 brain endothelial cell line. Images were acquired 1 day after seeding (HBVP) and 2 h after seeding (hCMEC/D3). (D) Representative images of low, high, and optimal seeding densities for hiPSC-derived brain pericyte-like cells. Images were acquired 1 day after seeding. (E) Representative images of final HBVP and hCMEC/D3 co-culture and hCMEC/D3 monoculture. Images were acquired 8 days following HBVP seeding and 7 days following hCMEC/D3 seeding. (F) Representative images of failed and successful BPLC and EECM-BMEC-like cell co-culture and EECM-BMEC-like cell monoculture. Images were acquired 7 days following BPLC seeding and 6 days following EECM-BMEC seeding. Underseeded BPLC cultures fail to have sufficient coverage, whereas overseeding BPLC cultures will grow overconfluent and start clumping/receding. Scale bars = 100 µm (C–F). Abbreviations: HBVPs = human brain vascular pericytes; hiPSC = human induced pluripotent stem cell; BPLCs = hiPSC-derived brain pericyte-like cells. Please click here to view a larger version of this figure.
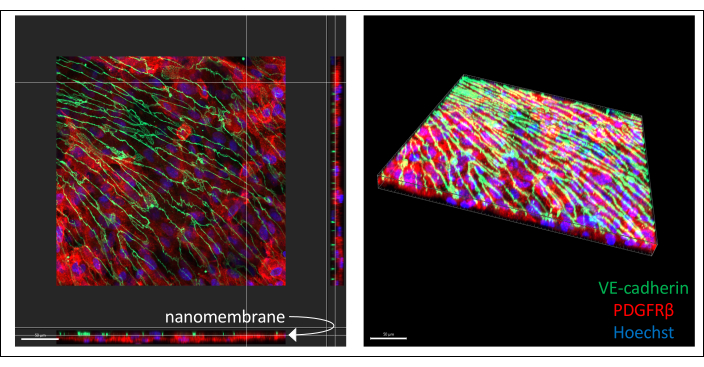
Figure 3: Representative images of immuno-stained hiPSC-derived cell co-culture in devices. Cells were stained for endothelial cell marker VE-cadherin (green), pericyte marker PDGFRβ (red), and nuclear stain (blue). Two layers of cells can be seen in close proximity, separated only by a thin silicon-nitride nanomembrane (white arrow marks membrane location on left image). Scale bar = 50 µm. Abbreviations: hiPSC = human induced pluripotent stem cell; PDGFRβ = platelet-derived growth factor receptor beta. Please click here to view a larger version of this figure.
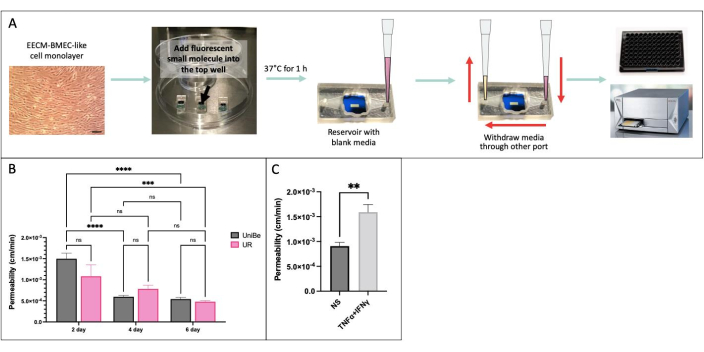
Figure 4: Sampling-based small molecule permeability assay. (A) Schematic of the experimental workflow. (B) Demonstration of the interlaboratory reproducibility between two physically distant laboratories at the University of Bern (UniBe), Switzerland, and the University of Rochester (UR), NY, USA: hiPSC-derived endothelial cells were cultured in the µSiM device for 2, 4, or 6 days and in transwell filters for 6 days. Permeability assay was performed using 150 µg/mL Lucifer Yellow (457 Da). The red bar indicates the previously published sodium fluorescein (376 Da) permeability data of the same hiPSC-derived endothelial cells cultured for 6 days in transwell filters40. N = 4-16 per group. Two-way ANOVA with Tukey's post hoc test was used, and comparisons were only displayed for relevant p < 0.05. (C) Demonstration of cytokine response using hiPSC-derived EECM-BMEC-like cells cultured in the µSiM for 2 days; 0.1 ng/mL TNFα + 2 IU/mL IFNγ) or media control (non-stimulated, NS) was added to the top chamber for 20 hr prior to permeability assay using 150 µg/mL Lucifer Yellow. N = 3 per group. Student’s t-test, p < 0.05. Please click here to view a larger version of this figure.
| Top Chamber Seeding Surface Area | Top Well Volume | Bottom Chamber Seeding Surface Area | Bottom Channel Volume |
| ~37 mm2 | 100 µL (can hold ≥115 µL) | ~42 mm2 | 10 µL (pipet 20 µL to avoid bubbles) |
Table 1: Critical µSiM surface area and volumes.
| Target | Fixative | Blocking Solution | Dilution |
| VE-cadherin | 4% PFA or 100% MeOH | 5% GS + 0.4% Tx-100 or 10% GS + 0.3% Triton X-100 | 1:50 |
| CD31 | 4% PFA or 100% MeOH | 5% GS + 0.3-0.4% Triton X-100 | 1:100 |
| Claudin-5 | 100% MeOH | 5-10% GS + 0.3% Triton X-100 | 1:200 |
| ZO-1 | 100% MeOH | 5-10% GS + 0.3% Triton X-100 | 1:200 |
| Occludin | 100% MeOH | 5-10% GS + 0.3% Triton X-100 | 1:50 |
| PDGFRβ | 4% PFA | 5% GS + 0.4% Triton X-100 | 1:100 |
| NG2 | 4% PFA | 5% GS + 0.4% Triton X-100 | 1:100 |
| Goat α-Mouse IgG Alexa Fluor 488 | 1:200 | ||
| Goat α-Mouse IgG Alexa Fluor 568 | 1:200 |
Table 2: Antibodies and staining methods validated for co-culture immunocytochemistry in µSiM devices. Abbreviations: PFA = paraformaldehyde; MeOH = methanol; GS = goat serum.
Supplemental File 1: Template for calculation of permeability value. Please click here to download this File.
