SLC genes can be cloned from RESOLUTE pDONR plasmids into BacMam vectors for mammalian expression
The described protocols for cloning, expression, and purification have proven successful for many SLC transporters across multiple protein folds. Nevertheless, the procedures include several checkpoints for monitoring progress, allowing for optimization to account for differences in expression, protein folding, lipid- and detergent-dependent stability, and sensitivity to buffer conditions.
Checkpoints during SLC cloning and small-scale expression
In the cloning steps, agarose gel electrophoresis should be used to ensure the correct size of the PCR and digestion products. Similarly, the Gateway and transposition reactions can be validated with a colony PCR reaction (Figure 2A,B). Baculovirus generation can be monitored using standard techniques as necessary49,50,51,52. The initial expression should be done at a small scale, evaluating protein yield by SDS-PAGE (Figure 2B). Similarly, the fraction of green fluorescent cells, total protein expression, and protein localization should be noted using fluorescence microscopy (Figure 2C,D). Protein expression should be optimized for cell type, temperature, time, and the necessity of co-expressing chaperones or complex partners. The expression can be further optimized by modifying the construct to truncate disordered N- and C-termini, based on secondary structure prediction56,57,58, and testing the types and placement of affinity tags. Protein stability should be evaluated at a small scale by FSEC (Figure 2E), SEC-based thermal shift assay (SEC-Ts), and DSF41,42,59,60,61,62. Small molecules, such as substrates and inhibitors, detergents, cholesterol hemisuccinate, lipids, and pH should be tested for improving protein stability considering the protein's function and native subcellular environment and subsequent purification buffers modified accordingly. In both small- and large-scale expression setups, cells should be monitored using microscopy for viability and contamination.
Optimization of transporter purification at large scale
Each step of large-scale protein purification should be evaluated by SDS-PAGE, including in-gel fluorescence to specifically monitor the GFP-tagged protein and enzymatic removal of that tag. In practice, the GFP-tagged SLC-expressing cells appear yellowish-green. After Twin-Strep-tag chromatographic elution, the eluent containing the purified protein appears fluorescent neon-green under white light. Chemically and structurally homogeneous protein should yield a single monodisperse A280 peak during size-exclusion chromatography (Figure 2F,G), and should show a single band on SDS-PAGE. The SDS-PAGE band corresponding to the expected SLC, and any unexpected bands, should be analyzed using tryptic digestion mass-spectrometry. Multiple bands on the SDS-PAGE gel indicate either proteolytic degradation, contaminating proteins, or SDS-resistant oligomers. Contaminating proteins may be removed by increasing the NaCl concentration of the solubilization buffer or changing the affinity tag. Proteolysis can be limited by improving the protein's purity, ensuring all steps are done at 4 °C or on ice, and optimizing the protocol to minimize the time of each step. If the SEC profile has a broad peak, multiple peaks, or large void peak (such as the purple trace of Figure 2F), the construct and purification conditions should be optimized at a small scale using FSEC, SEC-Ts, or DSF41,42,59,60,61,62.
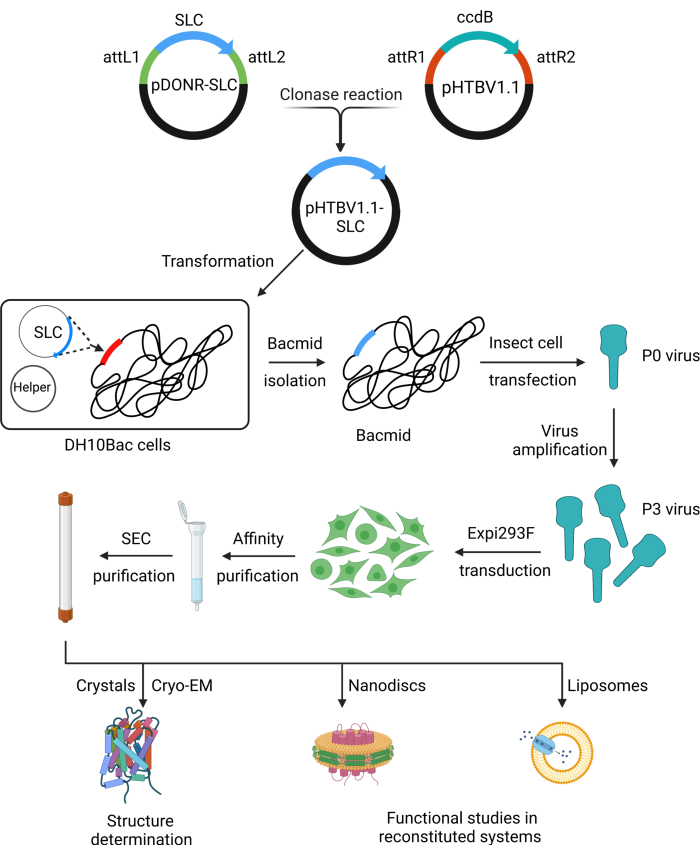
Figure 1: Schematic of RESOLUTE workflow for SLC expression and purification. Step-by-step illustration of recombination cloning, BacMam baculovirus preparation, protein expression and purification, and downstream applications. Abbreviations: SLC = solute carrier; Cryo-EM = cryo-electron microscopy; SEC = size exclusion chromatography. Please click here to view a larger version of this figure.
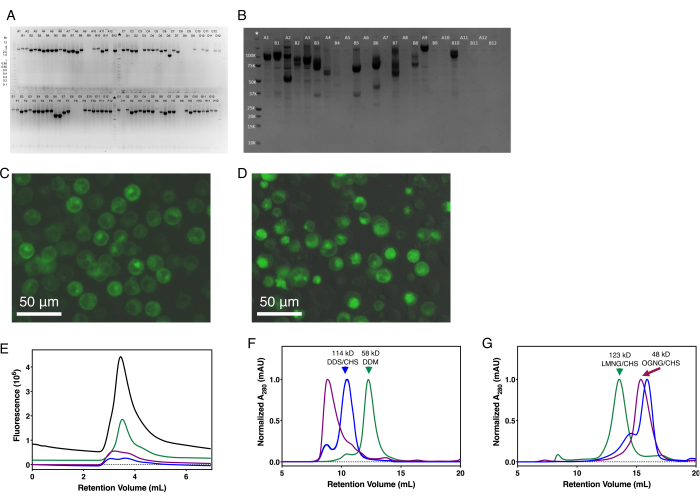
Figure 2: Representative results for SLC expression and purification. (A) Colony PCR of high-throughput SLC cloning into pHTBV1.1-C3CGFP-SIII-10H-GTW. (B) Coomassie-stained SDS-PAGE of single small-scale, parallel expression test of 24 different full-length SLCs. (C) In-cell fluorescence of a GFP-tagged SLC localizing primarily to the plasma membrane. (D) In-cell fluorescence of a GFP-tagged SLC with significant intracellular localization. (E) Representative FSEC traces for four SLCs resolved on a hydrophilic, neutral silica-based UHPLC column. Representative SEC traces for six SLCs purified on either a (F) dextran-agarose or (G) agarose size exclusion chromatography columns. Molecular weights of the SLC complex and detergent used for purification are indicated where the oligomeric state has been experimentally determined. Abbreviations: SLC = solute carrier; GFP = green fluorescent protein; FSEC = fluorescence-detection size exclusion chromatography. Please click here to view a larger version of this figure.
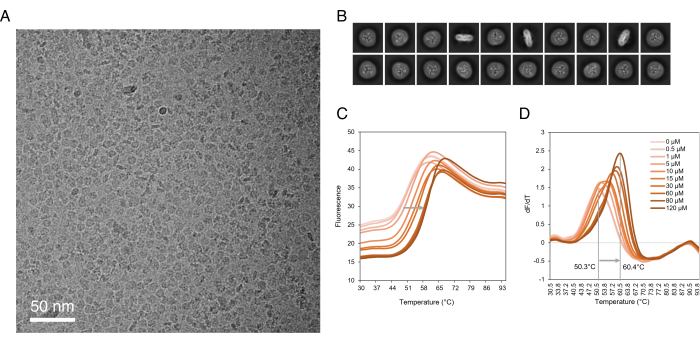
Figure 3: Downstream applications of purified SLCs. (A) Micrograph of SLC1A1 in detergent. (B) 2D class averages of SLC1A1 in detergent. (C) Raw fluorescence of CPM thermal denaturation assay of SLC10A6 incubated with various concentrations of Taurolithocholic acid 3-sulfate. (D) First derivative of CPM thermal denaturation of SLC10A6. The SLC10A6's melting temperature increased by 10 °C in the presence of 120 µM Taurolithocholic acid 3-sulfate. Abbreviations: SLC = solute carrier; CPM = N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide. Please click here to view a larger version of this figure.
| Vector Name | Antibiotic markers | Tags for Purification | Screening Primers | bp added during PCR | ||
| pHTBV1.1-C3CGFP-SIII-10H-GTW | AmpR | C-terminal | pHTBV-F (CTATAGACTCTATAGGCACACC) | ~350 | ||
| 3C protease site GFP | ||||||
| Twin-Strep | GFP-R (CTGTCGTACAGATGAACTTCAAGGTC) | |||||
| His10 | ||||||
| pDONR221 | KanR | none | M13 Forward | ~190 | ||
| M13 Reverse | ||||||
Table 1: Plasmids used for RESOLUTE cloning and BacMam generation.
| Solution | Composition |
| DH10Bac selection plates | LB-agar plates containing 50 µg/mL kanamycin, 10 µg/mL tetracycline, 7µg/mL gentamycin, 40 µg/mL IPTG, and 100 µg/mL bluo-gal |
| 2x LB medium (with antibiotics) | 2x LB broth containing 50 µg/mL kanamycin, 10 µg/mL tetracycline, and 7 µg/mL gentamycin |
| HT Lysis buffer | 50 mM HEPES-NaOH, pH 7.5, 250 mM NaCl, 5% glycerol, EDTA-free protease inhibitor |
| HT Wash buffer | 50 mM HEPES-NaOH, pH 7.5, 250 mM NaCl, 5% glycerol, 0.03% DDM/0.003% CHS |
| HT Elution buffer | 50 mM HEPES-NaOH, pH 7.5, 250 mM NaCl, 5% glycerol, 0.03% DDM/0.003% CHS, 100 mM D-Biotin |
| Base buffer | 50 mM HEPES-NaOH, pH 7.5, 150 mM NaCl |
| Detergent stock solution | 10% (w/v) detergent, with or without 1% CHS as appropriate. Mix at 4 °C overnight and store at -20 °C. |
| Strep wash buffer | 50 mM HEPES-NaOH, pH 7.5, 150 mM NaCl, detergent at 3-fold CMC, 10 mM MgCl2, 1 mM ATP |
| Strep elution buffer | 50 mM HEPES-NaOH, pH 7.5, 150 mM NaCl, 100 mM D-Biotin, detergent at 3-fold CMC |
| Size exclusion chromatography (SEC) buffer | 20 mM HEPES-NaOH, pH 7.5, 150 mM NaCl, detergent at 2-fold CMC. Filter through a 0.22 µM membrane. |
| Sodium butyrate | 1 M solution in DPBS, store at -20 °C for long-term use. |
Table 2: A list of solutions used in this protocol and their composition.
| Detergent system | Extraction concentration | Purification concentration (% w/v) |
| DDM | 1% | 0.03% |
| DDM + CHS | 1% + 0.1% | 0.03% + 0.003% |
| DM | 1% | 0.25% |
| DM+CHS | 1% + 0.1% | 0.25% + 0.025% |
| NG | 1% | 0.60% |
| OG | 1.5% | 1.50% |
| LDAO | 1% | 0.07% |
| LDAO + CHS | 1% + 0.1% | 0.07% + 0.007% |
| C12E8 | 1% | 0.015% |
| C12E8 + CHS | 1% + 0.1% | 0.015% + 0.0015% |
| C12E9 + CHS | 1% + 0.1% | 0.01 + 0.001% |
| CYMAL-5 | 1% | 0.40% |
| LMNG | 1% | 0.005% |
| LMNG + CHS | 1% + 0.1% | 0.005% + 0.0005% |
| GDN | 1% | 0.02% |
| Digitonin | 0.05% | |
| OGNG + CHS | 1% + 0.1% | 0.18% + 0.018% |
| C12E10 + CHS | 1% + 0.1% | 0.04% + 0.004% |
| Fos Choline-12 | 1% | 0.14% |
Table 3: Standard detergents used to test membrane solubilization and SLC monodispersity and stability.
