Eukaryotic SelO consists of an N-terminal mitochondrial targeting sequence, a kinase-like domain, and a highly conserved selenocysteine at the C-terminus of the protein23. This mitochondrial-resident enzyme encodes a pseudokinase domain that is conserved from bacteria to humans23. Structural analysis of the SelO homolog from Pseudomonas syringae revealed amino acid alterations in the active site that facilitate the binding of ATP in an inverted orientation in comparison to canonical kinases25. Accordingly, SelO catalyzes AMPylation, rather than phosphorylation, of multiple proteins involved in antioxidant signaling to protect cells from oxidative damage and cell death25. In addition to AMPylation, some prokaryotic and eukaryotic AMPylases have been shown to catalyze UMPylation27,28.
We used purified recombinant E. coli SelO in TSA to examine the binding of ATP and UTP nucleotides to SelO in the presence of divalent cations. Similar to canonical kinases, SelO harbors the DFG motif in which the aspartate binds the metal ion to coordinate nucleotide in the active site. First, we optimized the concentration of protein and dye for robust fluorescence measurements in TSA, determining that 5 µM to 10 µM E. coli SelO was optimal with 5x SYPRO Orange (Figure 2). Furthermore, we showed that the no protein, as well as the no dye controls, had low fluorescence signal that was not responsive to the temperature increase. We detected approximately +4 °C and +12 °C increases in the thermal stability of E. coli SelO in the presence of ATP-Mg2+ and ATP-Mn2+, respectively (Figure 3). However, we did not observe a shift in thermal stability upon incubation with UTP, indicating that E. coli SelO may not exhibit detectable binding to UTP.
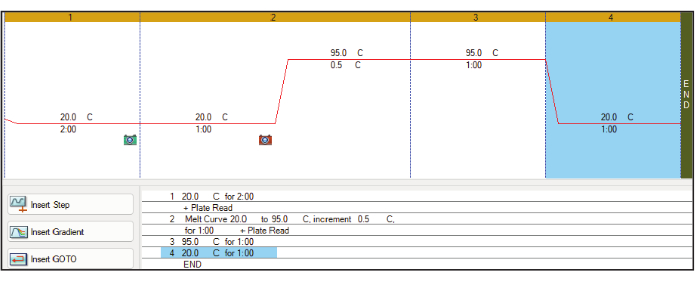
Figure 1: Thermocycler setup. An example of the thermocycler program used to increase sample temperature by +0.5 °C/min while measuring SYPRO Orange fluorescence using the FRET channel. The recommendation also includes an ambient temperature incubation at the start and end of the protocol. Abbreviation: FRET = Förster resonance energy transfer. Please click here to view a larger version of this figure.
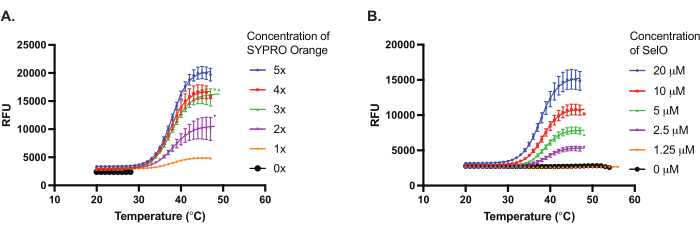
Figure 2: Optimization of protein and dye concentrations. (A) Representative results of optimizing the SYPRO Orange dye concentration using 5 µM Escherichia coli SelO. (B) Representative results of optimizing the E. coli SelO concentration using 5x SYPRO Orange dye. Thermal denaturation curve depicts relative fluorescence units (y-axis) with respect to temperature (x-axis). Results represent mean ± SD from triplicate wells (n = 3). Please click here to view a larger version of this figure.
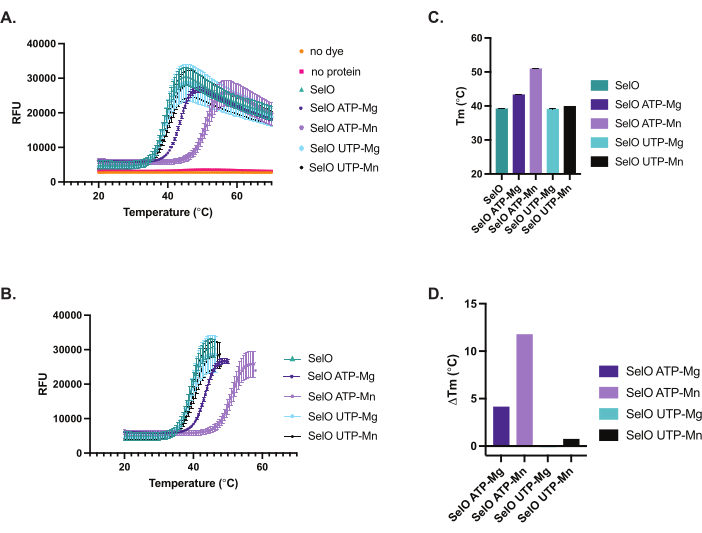
Figure 3:Thermal denaturation curves and ΔTm analyses of SelO in the presence of nucleotide and divalent cations. (A) Raw data of thermal denaturation curves of samples (5 µM Escherichia coli SelO in the presence of 200 µM ATP or UTP and 2 mM divalent cations, 5x SYPRO Orange) exported from RT-PCR machine and plotted. SelO ATP-Mg represents samples with SelO, ATP, and MgCl2, SelO ATP-Mn represents samples with SelO, ATP, and MnCl2, SelO UTP-Mg represents samples with SelO, UTP, and MgCl2, SelO UTP-Mn represents samples with SelO, UTP, and MnCl2. Note that the no dye and no protein controls exhibit low fluorescence. Results represent mean ± SD from triplicate wells (n = 3). (B) Thermal denaturation curves plotted to the highest intensity and fit with the Boltzmann sigmoidal equation. Although thermal denaturation of SelO was not affected by UTP, we observed a marked shift in thermal denaturation in the presence of ATP (purple curves). Results represent mean ± SD from triplicate wells (n = 3). (C) Histogram depicting Tm values as derived from Boltzmann sigmoidal fit of data represented in Figure 3B. Results represent mean ± SD from triplicate wells (n = 3). SelO 39.2 °C ± 0.085, SelO ATP-Mg 43.3 °C ± 0.133, SelO ATP-Mn 50.9 °C ± 0.085, SelO UTP-Mg 39.1 °C ± 0.056, SelO UTP-Mn 39.9 °C ± 0.087. (D) Histogram depicting the change in melting temperature ΔTm (Tm additive– Tm buffer). Please click here to view a larger version of this figure.
Supplemental Table S1: Example data for TSA of SelO in the presence of metals and nucleotides. Spreadsheet of raw data exported from the RT-PCR machine, and data processed to delete values after maximum RFU intensity as described in protocol steps 5.1 and 5.2. Please click here to download this File.
