Quantitative Detection of DNA-Protein Crosslinks and Their Post-Translational Modifications
Summary
The present protocol highlights a modified method to detect and quantify DNA-protein crosslinks (DPCs) and their post-translational modifications (PTMs), including ubiquitylation, SUMOylation, and ADP-ribosylation induced by topoisomerase inhibitors and by formaldehyde, thereby allowing the study of the formation and repair of DPCs and their PTMs.
Abstract
DNA-protein crosslinks (DPCs) are frequent, ubiquitous, and deleterious DNA lesions, which arise from endogenous DNA damage, enzyme (topoisomerases, methyltransferases, etc.) malfunctioning, or exogenous agents such as chemotherapeutics and crosslinking agents. Once DPCs are induced, several types of post-translational modifications (PTMs) are promptly conjugated to them as early response mechanisms. It has been shown that DPCs can be modified by ubiquitin, small ubiquitin-like modifier (SUMO), and poly-ADP-ribose, which prime the substrates to signal their respective designated repair enzymes and, in some cases, coordinate the repair in sequential manners. As PTMs transpire quickly and are highly reversible, it has been challenging to isolate and detect PTM-conjugated DPCs that usually remain at low levels. Presented here is an immunoassay to purify and quantitatively detect ubiquitylated, SUMOylated, and ADP-ribosylated DPCs (drug-induced topoisomerase DPCs and aldehyde-induced non-specific DPCs) in vivo. This assay is derived from the RADAR (rapid approach to DNA adduct recovery) assay that is used for the isolation of genomic DNA containing DPCs by ethanol precipitation. Following normalization and nuclease digestion, PTMs of DPCs, including ubiquitylation, SUMOylation, and ADP-ribosylation, are detected by immunoblotting using their corresponding antibodies. This robust assay can be utilized to identify and characterize novel molecular mechanisms that repair enzymatic and non-enzymatic DPCs and has the potential to discover small molecule inhibitors targeting specific factors that regulate PTMs to repair DPCs.
Introduction
Genomic DNA damage occurs due to spontaneous decay, internal damage, and environmental factors1. The resulting DNA lesions comprise damaged bases, mismatches, single- and double-strand breaks, inter- and intra-strand crosslinks, and DNA-protein crosslinks (DPCs). A DPC is formed when a chromatin-bound protein is trapped on DNA through covalent linkage. DPCs are induced by endogenous DNA lesions and reactive metabolites, as well as exogenous agents such as chemotherapeutics and bifunctional crosslinking agents. Under certain circumstances, enzyme dysfunction can also lead to the formation of DPCs2. The vast difference in DPC inducers results in a difference in the identity of the covalent-bound protein, the chromosome region where the DPC is formed, the structure type of the DNA crosslinked to the protein, and the chemical property of the covalent linkage between the protein and DNA2,3,4.
Based on their chemical nature, DPCs are generally categorized into two groups: enzymatic DPCs and non-enzymatic DPCs. Certain enzymes such as topoisomerases, glycosylases, and methyl/acyltransferases act by forming reversible enzyme-DNA covalent intermediates during their normal catalytic reactions. These are short-lived enzyme-DNA intermediates and can be converted into long-lived enzymatic DPCs upon their trapping by endogenous or exogenous agents, in particular by chemotherapeutics3. Topoisomerase DPCs are amongst the most frequent enzymatic DPCs in eukaryotic cells, which can be generated by clinically useful topoisomerase inhibitors (topotecan and irinotecan for topoisomerase I [TOP1] and etoposide and doxorubicin for topoisomerase II [TOP2]) and are the primary therapeutic mechanisms of these inhibitors5,6. DNA methyltransferases (DNMT) 1, 3A, and 3B are the target of 5-aza-2'-deoxycytidine (also known as decitabine) and form DPCs upon exposure to the drug7. Reactive agents, as well as ultraviolet light and ionizing radiation, induce non-enzymatic DPCs by non-specifically crosslinking proteins to DNA. Reactive aldehydes such as acetaldehyde and formaldehyde (FA) are often generated as byproducts of cellular metabolisms, among which FA is produced at micromolar concentrations during methanol metabolism, lipid peroxidation, and histone demethylation. Also, FA is a high-volume production chemical manufactured worldwide, to which many people are exposed both environmentally and occupationally8,9.
Both enzymatic and non-enzymatic DPCs are highly toxic to cells as their bulky protein components efficiently hinder nearly all chromatin-based processes, including replication and transcription, leading to cell cycle arrest and apoptosis if left unrepaired. Over the last two decades, the repair of DPCs has been vigorously studied, and several proteins/pathways have been identified as key factors that either directly repair DPCs or modulate their repair processes. For example, it has been well-established that proteolysis of the protein bulk of a DPC is a pivotal step of DPC repair, and that proteolysis can be catalyzed by the proteases SPRTN10,11,12,13,14, FAM111A15, GCNA16,17, or the 26S proteasome complex18,19,20,21,22,23,24,25,26,27 in a cell type- or cellular context-dependent manner. Identification and characterization of these proteases have largely relied on the in vivo complex of the enzyme (ICE) assay28,29 and the rapid approach to DNA adduct recovery (RADAR) assay30,31, both of which isolate DNA molecules and their covalent-bound proteins from free cellular proteins to allow the detection of DPCs by slot-blot using antibodies targeting the crosslinked proteins. Also, the trapped-in agarose DNA immunostaining (TARDIS) assay was used as a means of detecting and quantifying DPCs at the single-cell level32. Currently, researchers choose the RADAR assay over the ICE assay to measure DPCs, as the ICE assay relies on the purification of nucleic acids using cesium chloride gradient ultracentrifugation, which is extremely time-consuming, whereas the RADAR assay precipitates nucleic acids using ethanol within a much shorter period.
In recent years, mounting evidence has emerged that multiple post-translational modifications (PTMs) are involved in the signaling and recruitment of DPC-targeted proteases3,33,34,35. For example, both TOP1- and TOP2-DPCs were found to be conjugated by small ubiquitin-like modifier (SUMO)-2/3 and then SUMO-1 by the SUMO E3 ligase PIAS4, independently of DNA replication and transcription. The sequential SUMO modifications appear to be a target of ubiquitin, which is deposited to the SUMOylated TOP-DPCs and forms polymeric chains through its lysine 48 residue by a SUMO-targeted ubiquitin ligase termed RNF4. Subsequently, the ubiquitin polymer elicits a signal to and recruits the 26S proteasome to TOP-DPCs23,36. The same SUMO-ubiquitin pathway was recently shown to act on DNMT1-DPCs as well as PARP-DNA complexes for their repair37,38. In addition, SUMO-independent ubiquitylation by the ubiquitin E3 ligase TRAIP has been reported to prime DPCs for proteasomal degradation in a replication-coupled manner39. Akin to the proteasomal degradation of TOP-DPCs, proteolysis of enzymatic and non-enzymatic DPCs by the replication-coupled metalloprotease SPRTN also requires ubiquitylation of the DPC substrates as a mechanism to engage SPRTN40,41. Delineation of the role of SUMOylation and ubiquitylation requires the detection of DPCs that are marked with these PTMs. As the original ICE assay and RADAR assay rely on slot-blot/dot-blot apparatus to measure undigested DNA samples, neither of these two assays is able to resolve and visualize PTM-conjugated DPC species with different molecular weights. To overcome this problem, we digested the DNA samples following their purification by ethanol precipitation and sample normalization with micrococcal nuclease, a DNA and RNA endo-exonuclease to release the crosslinked proteins, which enabled us to resolve the proteins as well as their covalent PTMs with sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The electrophoresis allowed us to detect and quantitate PTM-conjugated DPCs using specific antibodies targeting the PTMs. We initially named this improved method the DUST assay, to highlight its robustness in the detection of ubiquitylated and SUMOylated TOP-DPCs23. Later, we expanded the use of the assay to quantitatively assess ADP-ribosylation of TOP1-DPCs in vivo, using antibodies against poly-ADP-ribose polymers20.
Presented here is a detailed protocol for the assay that detects and measures ubiquitylated, SUMOylated, and ADP-ribosylated DPCs, which was optimized for the modified TOP-DPCs that are induced by their inhibitors and non-specific/non-enzymatic DPCs that are induced by FA. This assay isolates PTM-conjugated DPCs by lysing cells with a chaotropic agent, precipitating DNA with ethanol, and releasing the otherwise crosslinked proteins and their modifiers with micrococcal nuclease. The otherwise DNA-bound proteins and their PTMs are quantified by immunoblotting using specific antibodies. This assay paves a new avenue to elucidate the molecular mechanisms by which the cell repairs both enzymatic and non-enzymatic DPCs. Specifically, it enables detailed studies of the induction and kinetics of PTMs important for the regulation of TOP-DPC degradation and repair, and thus permits the discovery of novel factors such as E3 ligases dictating the PTMs, as well as inhibitors targeting these factors. Since some of the PTMs responsible for TOP-DPC repair are likely involved in the repair of DPCs induced by other chemotherapeutics, such as platinum-based drugs22, this assay also has the potential for application to the discovery of new drugs and rational optimization of combinatorial therapies with topoisomerase inhibitors or platinum-based antineoplastics in patient cells to guide treatment regimens.
Protocol
1. Cell culture and drug treatment in the human embryonic kidney 293 (HEK293) cell line
- Prepare culture medium, Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum, 1% 2 mM L-glutamine, and 100 units/mL penicillin-streptomycin.
- Seed 1 x 106 cells in a 60 mm plate or a 6-well plate per treatment condition plus control.
- The next day, treat the cells with DPC inducers of choice.
- To induce TOP1-DPCs and their ubiquitylation and SUMOylation, add the TOP1 inhibitor camptothecin at 20 µM to the cells and collect the cells at 20, 60, and 180 min.
- To induce TOP2α and β-DPCs and their ubiquitylation and SUMOylation, expose the cells to add the TOP2 inhibitor etoposide at 200 µM to the cells and collect the cells at 20, 60, and 180 min.
- To induce non-enzymatic DPCs and their ubiquitylation and SUMOylation, add FA at 1 mM and collect the cells 2 h after the exposure.
- To induce the PARylation of TOP1-DPCs, pre-treat the cells for 1 h to block dePARylation with poly(ADP-ribose) glycohydrolase (PARG) inhibitor PDD00017273 at 10 µM, followed by co-treatment with 20 µM camptothecin for 20, 60, and 180 min.
2. Isolation and normalization of DNA containing crosslinked proteins
- Quickly aspirate the media with a suctioning pipette following treatment and rinse the cells with ice-cold 1x phosphate buffered saline (PBS). Immediately lyse the cells in 600 µL of DNAzol reagent containing 1x protease cocktail inhibitor, 1 mM dithiothreitol (DTT), and 20 mM N-ethylmaleimide (inhibitor of deSUMOylating and deubiquitylating enzymes).
- Slowly agitate the plate on a vibrating platform for 10 min at 4 °C.
- Add 1/2 volume of 100% cold ethanol (0.3 mL) directly to the plate and repeat step 2.2 until opaque nucleic acid aggregate becomes visible. Transfer the cell lysate to a 1.5 mL microcentrifuge tube and subject the tube to maximum speed (20,000 x g) centrifugation for 15 min at 4 °C to precipitate the nucleic acid and its crosslinked proteins.
- Aspirate the supernatant using a suctioning pipette and wash the nucleic acid pellet in 1 mL of 75% ethanol followed by 2 min of 20,000 x g centrifugation at 4 °C.
- Aspirate the supernatant, spin down at the same speed, and remove the remaining liquid using a P20 pipette. Air-dry the pellet for 5 min.
- Quickly dissolve the nucleic acid pellet in 0.1 mL of ddH2O. Resuspend the pellet by repeated pipetting then incubate in a 37 °C water bath until the pellet swells to at least three times bigger (approximately 30 min).
- Sonicate the samples with an ultrasonic processor probe at 30% amplitude for 10 s to fully dissolve the pellet.
- Optional step: Treat the samples with RNase A/T1 mix (10 µg of RNase A and 25 U of RNase T1) and incubate at 37 °C for 15 min. Add 1/10 volume of 3 M sodium acetate and two volumes of ice-cold 100% ethanol to the tube, followed by centrifugation at 20,000 x g for 10 min to retrieve the DNA. Remove the supernatant and dissolve the precipitated DNA in 0.1 mL of ddH2O.
- Optional step: Centrifuge the sample for 5 min at 20,000 x g and transfer the supernatant to a new tube.
- Quantify the DNA concentration using an ultraviolet-visible (UV-Vis) spectrometer. The typical DNA yield is around 600-800 ng/µL. The A260/A280 ratio should be reduced from 2.0-2.1 to 1.8-1.9 after RNA removal.
- Adjust the concentration of the DNA to 400-500 ng/µL in 0.12 mL of ddH2O. Transfer 20 µL of the sample to a new microcentrifuge tube as undigested DNA loading control (refer to step 2.4).
- To digest the DNA dissolved in the remaining 100 µL of ddH2O, add 2,000 gel units of micrococcal nuclease along with 1/10 volume (~11 µL) of 10x calcium micrococcal nuclease reaction buffer to the sample. Incubate at 37 °C for 30 min.
3. Western blotting of digested DNA samples
- Add 4x Laemmli sample buffer, then boil the sample for 5 min.
- Load 5-6 µg of digested sample (~15 µL) onto 4%-20% polyacrylamide gel, followed by SDS-PAGE42 to resolve unmodified and PTM-conjugated DPCs.
- To detect FA-induced non-enzymatic DPC species, incubate the gel with Coomassie blue stain overnight at room temperature. Wash the gel with ddH2O for 2 h and acquire an image using an imaging system.
- Transfer the gel and incubate the membranes overnight at 4 °C with appropriate dilutions of primary antibody in blocking buffer.
- To detect ubiquitylation, make a 1:100 dilution of anti-ubiquitin antibody.
- To detect SUMO-1 or SUMO-2/3 modification, make a 1:250 dilution of anti-SUMO-1 or anti-SUMO-2/3 antibody.
- To detection ADP-ribosylation, make a 1:500 dilution of anti-PAR antibody.
- To detect total TOP1-, TOP2α-, or TOP2β-DPCs, make a 1:500 dilution of anti-TOP1, anti-TOP2α or anti-TOP2β antibody.
NOTE: Refer to the Table of Materials for details on antibody dilution.
- Incubate a 1x PBS-T (0.1% tween 20) washed membrane with a secondary antibody diluted 5,000-fold in blocking buffer for 60 min at room temperature.
- Develop the membrane with enhanced chemiluminescence (ECL) reagent and acquire an image using the imaging system.
4. Slot-blotting of undigested DNA samples
- Dilute the 20 µL undigested DNA sample in 180 µL of sodium phosphate buffer (25 mM, pH 6.6).
- Cut the nitrocellulose membrane (0.45 µm) and equilibrate for 5 min in the sodium phosphate buffer.
- Assemble the slot-blot apparatus according to the manufacturer's instructions and connect it to a vacuum system.
- Wash the wells with sodium phosphate buffer by applying the vacuum. Make sure there is no leaking of the wells.
- Stop the vacuum and load 200 µL of DNA per sample (1 µg). Fill the empty wells with 200 µL of sodium phosphate buffer.
- Apply the vacuum.
- When all the wells are completely empty, stop the vacuum, load 200 µL of sodium phosphate buffer in each well, and repeat step 4.6.
- Retrieve the membrane and block with 5% blocking buffer for 0.5 h at room temperature.
- Probe with anti-double strand DNA (dsDNA) antibody at a 1:5,000 dilution overnight at 4 °C.
- Wash 3x with 1x PBS-T and incubate with 1:5,000 diluted horseradish peroxidase (HRP)-linked anti-mouse secondary antibody.
- Develop the membrane with enhanced chemiluminescence (ECL) reagent and acquire an image using the imaging system.
5. Densitometric analysis
- Using ImageJ, calculate the ratio of the intensity of each band/smear relative to the intensity of the slot of undigested DNA and normalize the ratio to that of cells without/before drug treatment.
Representative Results
The representative results presented in Figure 1 show the formation and kinetics of drug-induced TOP1-DPCs and their SUMOylation and ubiquitylation. TOP1 cleaved one strand of the DNA duplex and formed an enzyme-DNA covalent intermediate, termed TOP1 cleavage complex (TOP1cc). The treatment of camptothecin (CPT), a TOP1 inhibitor, bound to and stabilized TOP1cc, leading to the formation of long-life TOP1-DPCs. TOP1-DPCs were observed to be induced and peak 20 min following exposure to CPT. Concurrently, TOP1-DPCs were modified by SUMO-2/3, which also peaked 20 min after CPT treatment. As SUMO-2 and SUMO-3 share 95% sequence identity, the antibody does not distinguish one from the other. At 60 min, TOP1-DPCs and their SUMO-2/3 modification diminished, accompanied by the culmination of their SUMO-1 modification and ubiquitylation. After the 60 min drug treatment, the levels of TOP1-DPC SUMO-1 modification and ubiquitylation began to decline. In mammals, TOP2 isozymes α and β act by introducing a DNA double-strand break, as well as via the formation of a transient and reversible enzyme-DNA covalent complex (TOP2cc). TOP2 inhibitors, such as etoposide (ETOP), convert TOP2cc to TOP2-DPCs and induce their SUMOylation and ubiquitylation. Akin to the kinetics of TOP1-DPCs and their PTMs, TOP2α- and β-DPCs and their SUMO-2/3 modification reached a peak at 20 min, then started to decrease; meanwhile, their SUMO-1 and ubiquitin modifications peaked at 60 min (Figure 2). The clearance of TOP-DPCs has been demonstrated to result from proteasomal degradation, and the clearance of TOP-DPC SUMOylation and ubiquitylation is likely due to recycling by deSUMOylation and deubiquitylation, respectively, by their reversing enzymes. The experiments in Figure 3 examined FA-induced non-enzymatic DPCs and their PTMs. It was observed that the DPCs and their SUMO-2/3, SUMO-1, and ubiquitylation formed and accumulated in a FA dose-dependent manner. Finally, the PARylation of TOP1-DPCs was quantitively detected with an anti-PAR antibody using the same method (Figure 4). TOP1-DPC PARylation was not detectable unless a PARG inhibitor was added to the cell, suggesting that PARylation transpires promptly and is highly dynamic. Consistent with the previous finding, the inhibition of dePARylation by PARGi appeared to accumulate TOP1-DPCs, likely by blocking proteolytic degradation.
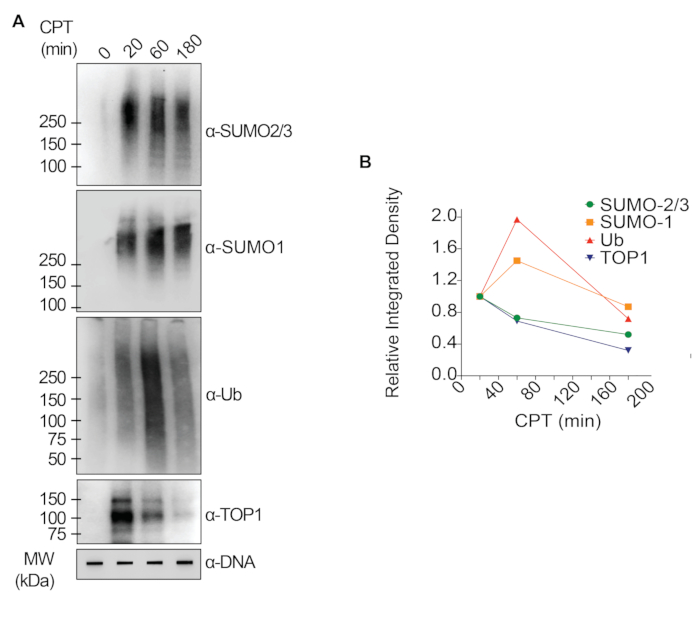
Figure 1: Quantitative analyses of formation and kinetics of TOP1-DPCs and their SUMOylation and ubiquitylation upon CPT treatment in HEK293 cells. (A) HEK293 cells were treated with 20 µM CPT for indicated periods of time. Cell lysates were harvested and subjected to the modified RADAR assay and western blotting with indicated antibodies. Undigested DNA samples were subjected to slot-blotting using anti-dsDNA antibody as a loading control. (B) Band intensities were quantified with ImageJ software and plotted with Prism software. Please click here to view a larger version of this figure.
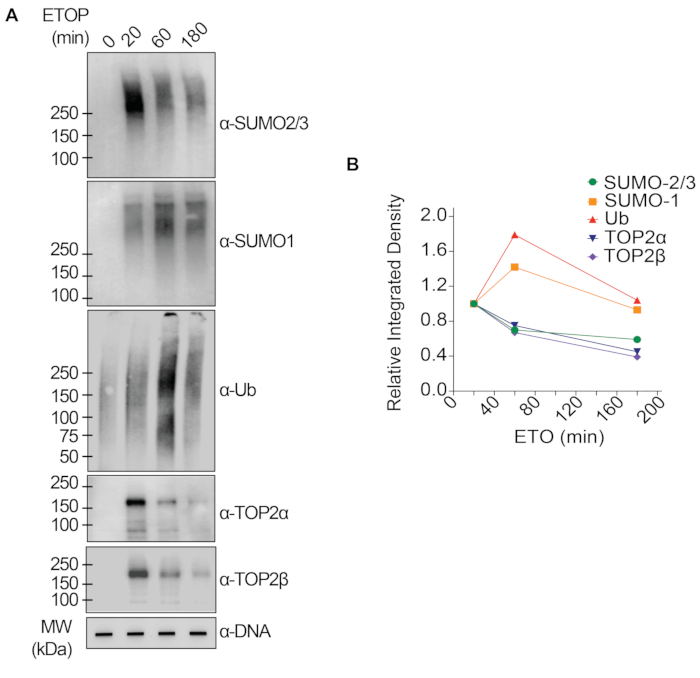
Figure 2: Quantitative analyses of formation and kinetics of TOP2-DPCs and their SUMOylation and ubiquitylation upon ETOP treatment in HEK293 cells. (A) HEK293 cells were treated with 200 µM ETOP for indicated periods of time. Cell lysates were harvested and subjected to the modified RADAR assay and western blotting with indicated antibodies. Undigested DNA samples were subjected to slot-blotting using anti-dsDNA antibody as a loading control. (B) Band intensities were quantified with ImageJ software and plotted with Prism software. Please click here to view a larger version of this figure.
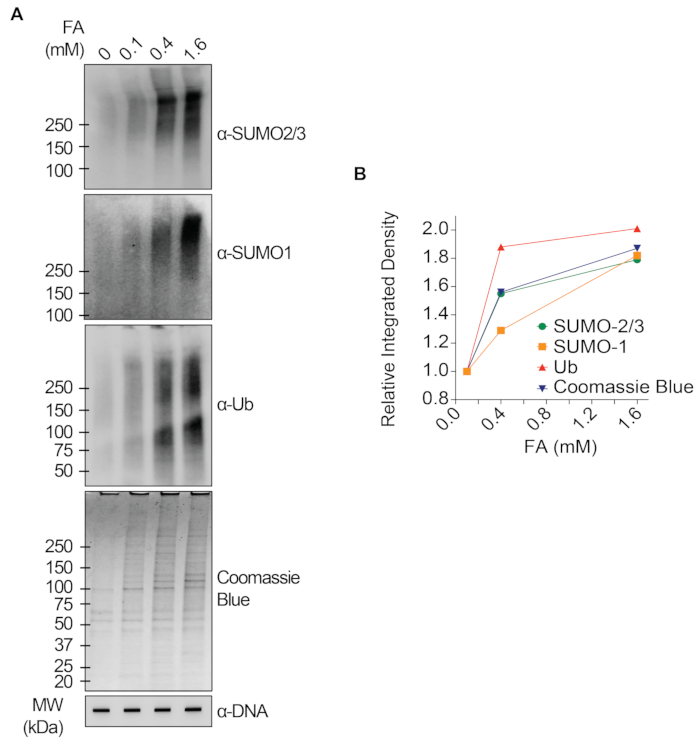
Figure 3: Quantitative analyses of non-enzymatic DPCs and their SUMOylation and ubiquitylation upon FA treatment in HEK293 cells. (A) HEK293 cells were treated with FA of indicated concentrations for 2 h. Cell lysates were harvested and subjected to the modified RADAR assay and western blotting with indicated antibodies. Undigested DNA samples were subjected to slot-blotting using anti-dsDNA antibody as a loading control. (B) Band intensities were quantified with ImageJ software and plotted with Prism software. Please click here to view a larger version of this figure.
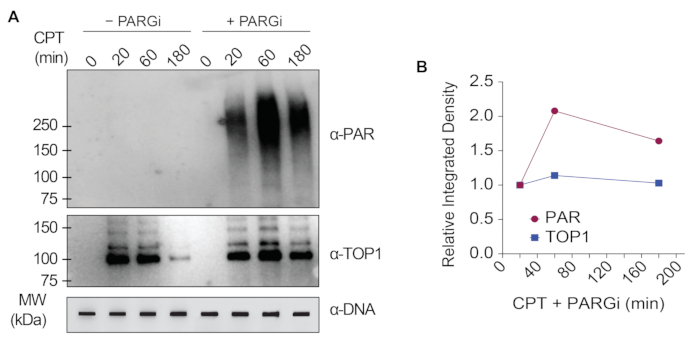
Figure 4: Quantitative analyses of TOP1-DPCs and their PARylation upon CPT treatment in HEK293 cells. (A) HEK293 cells were pre-treated with 10 µM PARGi for 1 h and then co-treated with CPT for indicated periods of time. Cell lysates were harvested and subjected to the modified RADAR assay and western blotting with indicated antibodies. Undigested DNA samples were subjected to slot-blotting using anti-dsDNA antibody as a loading control. (B) Band intensities were quantified with ImageJ software and plotted with Prism software. Please click here to view a larger version of this figure.
Discussion
The described method allows the measurement of enzymatic and non-enzymatic DNA-protein crosslinks in mammalian cells and is the only suitable approach for studying their ubiquitylation, SUMOylation, and ADP-ribosylation. Slot-blotting following the ICE or RADAR assay permits the fast detection of specific enzymatic DPCs such as TOP-DPCs using their antibodies. However, a caveat to this method is its inability to separate proteins of different molecular weights, making it impossible to determine the sizes of PTM-conjugated DPCs. The described method solves the problem by releasing crosslinked proteins with micrococcal nuclease, which degrades DNA to oligonucleotides with terminal 3'-phosphates, thereby permitting full separation of the proteins (conjugated with oligonucleotides) by SDS-PAGE. DPCs modified with ubiquitin, SUMO, or ADP-ribose monomers and polymers of different sizes can therefore be visualized and quantified by antibodies targeting these PTMs, enabling detailed investigation of their formation and kinetics. To ensure reproducibility and calculate statistical significance, biological replicates of the experiments are required.
One of the most common problems of this assay is low DNA yield after ethanol precipitation. On one hand, DNA yield can be increased with more starting material (cells). On the other hand, incubating cell lysates with ethanol in a flat plate rather than an Eppendorf tube can markedly improve the aggregation of DNA molecules and thus facilitate their precipitation. Non-specific signals observed in samples without drug treatments may indicate non-covalent protein contamination. If this is the case, one may consider washing DNA pellets with high-salt buffer to remove the contaminants prior to sonication. It is also recommended to spin down DNA samples after sonication and micrococcal nuclease digestion and discard any insoluble. In the case of poor or no signal, several potential solutions can be attempted. First, one can increase the loading amount of DNA for SDS-PAGE and immunoblotting. To make SUMOylated and ubiquitylated DPC species detectable, it is recommended to load at least 4 µg of DNA onto the gel. Second, one can increase the drug concentrations to induce higher levels of DPCs and their associated PTMs. Third, it is suggested to incubate blots with primary antibodies for another day if bands/smears appear to be weak. A 2-day incubation can significantly potentiate the signal and thus reduces biological variability from independent experiments23. Membrane stripping for re-staining inevitably results in the loss of a certain amount of PTM-conjugated DPC species that are already in low abundance. Therefore, it is highly recommended to run separate gels for ubiquitin and SUMO detection rather than re-probing one blot. In addition, DNA pellets must be washed with 75% ethanol to remove the remaining DNAzol containing guanidine salt before dissolution in H2O or any other solvents, which otherwise causes crystallization of the sample after the addition of Laemmli loading buffer.
The workflow of the described method is much more time-efficient compared to the cumbersome ICE assay, as it relies on fast ethanol precipitation instead of the time-consuming cesium chloride ultracentrifugation to isolate genomic DNA. At a price, ethanol-based purification brings about a low amount of protein contaminants that are normally negligible for immunodetection. However, when it comes to analytic studies, such as mass spectrometry-based proteomic analysis or next-generation sequencing that require accuracy and precision, cesium-chloride density-gradient centrifugation is still a more reliable approach for isolating pure, high-abundance DNA. This method can also potentially be applied to the profiling of modification sites on crosslinked proteins and the determination of linkage types of poly-ubiquitylation and poly-SUMOylation using proper mass spectrometry-based methods.
Of note, this assay allows the identification and characterization of factors regulating PTMs for DPCs repair. For instance, unbiased high-throughput screening methods (RNA interference and CRISPR) are powerful tools to discover ubiquitin E3 ligases, SUMO E3 ligases, and their associated cofactors mitigating cytotoxicity of DPC inducers. The described method enables molecular validation of these proteins by determining if they help cells survive DPC inducers by repairing the DPCs. Novel small-molecule inhibitors targeting these proteins identified, for example, by virtual screening, can also be validated using this protocol. Given that topoisomerase inhibitors are among the most prescribed chemotherapeutics, this robust assay can be developed as a tool for the development of drugs that synergize with clinical topoisomerase inhibitors.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was in part supported by National Cancer Institute Center for Cancer ResearchExcellence in Postdoctoral Research Transition Award.
Materials
| 10x Phosphate buffered saline (PBS) | Thermo Fisher | 70011069 | |
| 4–20% precast polyacrylamide gel | Bio-Rad | 4561096 | |
| 4x Laemmli Sample Buffer | Bio-Rad | 1610747 | |
| AcquaStain (coomassie blue) | Bulldog Bio | AS001000 | |
| anti-dsDNA (mouse monoclonal) | Abcam | 27156 | 1: 5,000 dilution is recommended |
| anti-PAR (mouse monoclonal) | R&D systems | 4335-MC-100 | 1: 500 dilution is recommended |
| anti-SUMO-1(rabbit monoclonal) | Cell Signaling Technology | 4940 | 1: 250 dilution is recommended |
| anti-SUMO-2/3 (rabbit monoclonal) | Cell Signaling Technology | 4971 | 1: 250 dilution is recommended |
| anti-TOP1 (mouse monoclonal) | BD Biosciences | 556597 | 1: 500 dilution is recommended |
| anti-TOP2α (mouse monoclonal) | Santa Cruz Biotechnology | SC-365799 | 1: 250 dilution is recommended |
| anti-TOP2β (mouse monoclonal) | Santa Cruz Biotechnology | SC-25330 | 1: 250 dilution is recommended |
| anti-ubiquitin (mouse monoclonal) | Santa Cruz Biotechnology | SC-8017 | 1: 100 dilution is recommended |
| Calcium chloride | Sigma-Aldrich | 499609 | Used for micrococcal nuclease digestion |
| Camptothecin | Sigma-Aldrich | PHL89593 | |
| ChemiDo MP imaging system | Bio-Rad | 12003154 | |
| Disodium phosphate | Sigma-Aldrich | 5438380100 | Used to make sodium phosphate buffer |
| DNAzol | Thermo Fisher | 10503027 | |
| DTT (dithiothreitol) | Thermo Fisher | R0861 | |
| Dulbecco's modified eagle's medium | Sigma-Aldrich | 11965084 | |
| Ethyl alcohol, 200 proof | Sigma-Aldrich | E7023 | |
| Etoposide | Sigma-Aldrich | 1268808 | |
| Formaldehyde | Sigma-Aldrich | 47608 | |
| Graphpad Prism Software | GraphStats | Prism 9.0.0 | |
| HRP-linked Mouse IgG | Cytiva | NA931 | 1: 5,000 dilution is recommended |
| HRP-linked Rabbit IgG | Cytiva | NA934 | 1: 5,000 dilution is recommended |
| ImageJ Software | NIH, USA | ImageJ 1.53e | |
| L-Glutamine | Fisher Scientific | 25030081 | |
| Maximum sensitivity ECL substrate | Thermo Fisher | 34095 | |
| Micrococcal nuclease | New England BioLabs | M0247S | |
| Monosodium phosphate | Sigma-Aldrich | S3139 | Used to make sodium phosphate buffer |
| NanoDrop 2000 spectrophotometer | Thermo Scientific | ND-2000 | |
| N-ethylmaleimide | Thermo Fisher | 23030 | DeSUMOylation/deubiquitylation inhibitor |
| Nitrocellulose membrane, 0.45 µm | Bio-Rad | 1620115 | |
| Non-fat dry milk | Bio-Rad | 1706404XTU | |
| PDD00017273 | Selleckchem | S8862 | Poly(ADP-ribose) glycohydrolase inhibitor |
| Penicillin-Streptomycin | Thermo Fisher | 15140122 | |
| Protease inhibitor cocktail | Thermo Fisher | 78430 | |
| Q700 sonicator | Qsonica | Q700-110 | |
| Ready-to-assemble PVDF transfer kit | Bio-Rad | 1704274 | |
| Slot-blot apparatus | Bio-Rad | 1706542 | |
| Slot-blot filter paper | Bio-Rad | 1620161 | |
| Trans-Blot turbo transfer system | Bio-Rad | 1704150 | |
| Tris/Glycine/SDS electrophoresis buffer | Bio-Rad | 1610732 | |
| Tween-20 | Sigma-Aldrich | P3179 | |
| Vertical electrophoresis cell | Bio-Rad | 1658004 |
References
- Jackson, S. P., Bartek, J. The DNA-damage response in human biology and disease. Nature. 461 (7267), 1071-1078 (2009).
- Klages-Mundt, N. L., Li, L. Formation and repair of DNA-protein crosslink damage. Science China. Life Sciences. 60 (10), 1065-1076 (2017).
- Weickert, P., Stingele, J. DNA-protein crosslinks and their resolution. Annual Review of Biochemistry. 91, 157-181 (2022).
- Tretyakova, N. Y., Groehler, A., Ji, S. DNA-Protein cross-links: formation, structural identities, and biological outcomes. Accounts of Chemical Research. 48 (6), 1631-1644 (2015).
- Pommier, Y., Nussenzweig, A., Takeda, S., Austin, C. Human topoisomerases and their roles in genome stability and organization. Nature Reviews Molecular Cell Biology. 23 (6), 407-427 (2022).
- Pommier, Y., Sun, Y., Huang, S. N., Nitiss, J. L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nature Reviews Molecular Cell Biology. 17 (11), 703-721 (2016).
- Ruggiano, A., Ramadan, K. DNA-protein crosslink proteases in genome stability. Communications Biology. 4 (1), 11 (2021).
- Hoffman, E. A., Frey, B. L., Smith, L. M., Auble, D. T. Formaldehyde crosslinking: a tool for the study of chromatin complexes. The Journal of Biological Chemistry. 290 (44), 26404-26411 (2015).
- Moretton, A., Loizou, J. I. Interplay between cellular metabolism and the DNA damage response in cancer. Cancers. 12 (8), 2051 (2020).
- Stingele, J., Schwarz, M. S., Bloemeke, N., Wolf, P. G., Jentsch, S. A DNA-dependent protease involved in DNA-protein crosslink repair. Cell. 158 (2), 327-338 (2014).
- Maskey, R. S., et al. Spartan deficiency causes accumulation of topoisomerase 1 cleavage complexes and tumorigenesis. Nucleic Acids Research. 45 (8), 4564-4576 (2017).
- Stingele, J., et al. Mechanism and regulation of DNA-protein crosslink repair by the DNA-dependent metalloprotease SPRTN. Molecular Cell. 64 (4), 688-703 (2016).
- Vaz, B., et al. Metalloprotease SPRTN/DVC1 orchestrates replication-coupled DNA-protein crosslink repair. Molecular Cell. 64 (4), 704-719 (2016).
- Lopez-Mosqueda, J., et al. SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks. eLife. 5, e21491 (2016).
- Kojima, Y., et al. FAM111A protects replication forks from protein obstacles via its trypsin-like domain. Nature Communications. 11 (1), 1318 (2020).
- Dokshin, G. A., et al. GCNA interacts with spartan and topoisomerase II to regulate genome stability. Developmental Cell. 52 (1), 53-68 (2020).
- Borgermann, N., et al. SUMOylation promotes protective responses to DNA-protein crosslinks. The EMBO Journal. 38 (8), e101496 (2019).
- Sun, Y., Zhang, Y., Schultz, C. W., Pommier, Y., Thomas, A. CDK7 inhibition synergizes with topoisomerase I inhibition in small cell lung cancer cells by inducing ubiquitin-mediated proteolysis of RNA polymerase II. Molecular Cancer Therapeutics. 21 (9), 1430-1438 (2022).
- Sun, Y., et al. Requirements for MRN endonuclease processing of topoisomerase II-mediated DNA damage in mammalian cells. Frontiers in Molecular Biosciences. 9, 1007064 (2022).
- Sun, Y., et al. PARylation prevents the proteasomal degradation of topoisomerase I DNA-protein crosslinks and induces their deubiquitylation. Nature Communications. 12 (1), 5010 (2021).
- Sun, Y., et al. Excision repair of topoisomerase DNA-protein crosslinks (TOP-DPC). DNA Repair. 89, 102837 (2020).
- Sun, Y., Saha, L. K., Saha, S., Jo, U., Pommier, Y. Debulking of topoisomerase DNA-protein crosslinks (TOP-DPC) by the proteasome, non-proteasomal and non-proteolytic pathways. DNA Repair. 94, 102926 (2020).
- Sun, Y., et al. A conserved SUMO pathway repairs topoisomerase DNA-protein cross-links by engaging ubiquitin-mediated proteasomal degradation. Science Advances. 6 (46), (2020).
- Saha, S., et al. DNA and RNA cleavage complexes and repair pathway for TOP3B RNA- and DNA-protein crosslinks. Cell Reports. 33 (13), 108569 (2020).
- Swan, R. L., Cowell, I. G., Austin, C. A. Mechanisms to repair stalled topoisomerase II-DNA covalent complexes. Molecular Pharmacology. 101 (1), 24-32 (2022).
- Swan, R. L., Poh, L. L. K., Cowell, I. G., Austin, C. A. Small molecule inhibitors confirm ubiquitin-dependent removal of TOP2-DNA covalent complexes. Molecular Pharmacology. 98 (3), 222-233 (2020).
- Sciascia, N., et al. Suppressing proteasome mediated processing of topoisomerase II DNA-protein complexes preserves genome integrity. eLife. 9, e53447 (2020).
- Anand, J., Sun, Y., Zhao, Y., Nitiss, K. C., Nitiss, J. L. Detection of topoisomerase covalent complexes in eukaryotic cells. Methods in Molecular Biology. 1703, 283-299 (2018).
- Subramanian, D., Furbee, C. S., Muller, M. T. ICE bioassay. Isolating in vivo complexes of enzyme to DNA. Methods in Molecular Biology. 95, 137-147 (2001).
- Nitiss, J. L., Kiianitsa, K., Sun, Y., Nitiss, K. C., Maizels, N. Topoisomerase assays. Current Protocols. 1 (10), e250 (2021).
- Kiianitsa, K., Maizels, N. A rapid and sensitive assay for DNA-protein covalent complexes in living cells. Nucleic Acids Research. 41 (9), e104 (2013).
- Cowell, I. G., Tilby, M. J., Austin, C. A. An overview of the visualisation and quantitation of low and high MW DNA adducts using the trapped in agarose DNA immunostaining (TARDIS) assay. Mutagenesis. 26 (2), 253-260 (2011).
- Leng, X., Duxin, J. P. Targeting DNA-protein crosslinks via post-translational modifications. Frontiers in Molecular Biosciences. 9, 944775 (2022).
- Kuhbacher, U., Duxin, J. P. How to fix DNA-protein crosslinks. DNA Repair. 94, 102924 (2020).
- Stingele, J., Bellelli, R., Boulton, S. J. Mechanisms of DNA-protein crosslink repair. Nature Reviews Molecular Cell Biology. 18 (9), 563-573 (2017).
- Sun, Y., Nitiss, J. L., Pommier, Y. SUMO: A Swiss army knife for eukaryotic topoisomerases. Frontiers in Molecular Biosciences. 9, 871161 (2022).
- Krastev, D. B., et al. The ubiquitin-dependent ATPase p97 removes cytotoxic trapped PARP1 from chromatin. Nature Cell Biology. 24 (1), 62-73 (2022).
- Liu, J. C. Y., et al. Mechanism and function of DNA replication-independent DNA-protein crosslink repair via the SUMO-RNF4 pathway. The EMBO Journal. 40 (18), e107413 (2021).
- Larsen, N. B., et al. Replication-coupled DNA-protein crosslink repair by SPRTN and the proteasome in Xenopus egg extracts. Molecular Cell. 73 (3), 574-588 (2019).
- Zhao, S., et al. A ubiquitin switch controls autocatalytic inactivation of the DNA-protein crosslink repair protease SPRTN. Nucleic Acids Research. 49 (2), 902-915 (2021).
- Ruggiano, A., et al. The protease SPRTN and SUMOylation coordinate DNA-protein crosslink repair to prevent genome instability. Cell Reports. 37 (10), 110080 (2021).
- Mahmood, T., Yang, P. C. Western blot: technique, theory, and trouble shooting. North American Journal of Medical Sciences. 4 (9), 429-434 (2012).
