Using Live Cell STED Imaging to Visualize Mitochondrial Inner Membrane Ultrastructure in Neuronal Cell Models
Summary
This protocol presents a workflow for the propagation, differentiation, and staining of cultured SH-SY5Y cells and primary rat hippocampal neurons for mitochondrial ultrastructure visualization and analysis using stimulated emission depletion (STED) microscopy.
Abstract
Mitochondria play many essential roles in the cell, including energy production, regulation of Ca2+ homeostasis, lipid biosynthesis, and production of reactive oxygen species (ROS). These mitochondria-mediated processes take on specialized roles in neurons, coordinating aerobic metabolism to meet the high energy demands of these cells, modulating Ca2+ signaling, providing lipids for axon growth and regeneration, and tuning ROS production for neuronal development and function. Mitochondrial dysfunction is therefore a central driver in neurodegenerative diseases. Mitochondrial structure and function are inextricably linked. The morphologically complex inner membrane with structural infolds called cristae harbors many molecular systems that perform the signature processes of the mitochondrion. The architectural features of the inner membrane are ultrastructural and therefore, too small to be visualized by traditional diffraction-limited resolved microscopy. Thus, most insights on mitochondrial ultrastructure have come from electron microscopy on fixed samples. However, emerging technologies in super-resolution fluorescence microscopy now provide resolution down to tens of nanometers, allowing visualization of ultrastructural features in live cells. Super-resolution imaging therefore offers an unprecedented ability to directly image fine details of mitochondrial structure, nanoscale protein distributions, and cristae dynamics, providing fundamental new insights that link mitochondria to human health and disease. This protocol presents the use of stimulated emission depletion (STED) super-resolution microscopy to visualize the mitochondrial ultrastructure of live human neuroblastoma cells and primary rat neurons. This procedure is organized into five sections: (1) growth and differentiation of the SH-SY5Y cell line, (2) isolation, plating, and growth of primary rat hippocampal neurons, (3) procedures for staining cells for live STED imaging, (4) procedures for live cell STED experiments using a STED microscope for reference, and (5) guidance for segmentation and image processing using examples to measure and quantify morphological features of the inner membrane.
Introduction
Mitochondria are eukaryotic organelles of endosymbiotic origin that are responsible for regulating several key cellular processes, including intermediary metabolism and ATP production, ion homeostasis, lipid biosynthesis, and programmed cell death (apoptosis). These organelles are topologically complex, containing a double membrane system that establishes multiple subcompartments1 (Figure 1A). The outer mitochondrial membrane (OMM) interfaces with the cytosol and establishes direct inter-organelle contacts2,3. The inner mitochondrial membrane (IMM) is an energy-conserving membrane that maintains ion gradients stored primarily as an electric membrane potential (ΔΨm) to drive ATP synthesis and other energy-requiring processes4,5. The IMM is further subdivided into the inner boundary membrane (IBM), which is closely appressed to the OMM, and protruding structures called cristae that are bound by the cristae membrane (CM). This membrane delineates the innermost matrix compartment from the intracristal space (ICS) and the intermembrane space (IMS).
Mitochondria have a dynamic morphology based on continuous and balanced processes of fission and fusion that are governed by mechanoenzymes of the dynamin superfamily6. Fusion allows for increased connectivity and formation of reticular networks, whereas fission leads to mitochondrial fragmentation and enables the removal of damaged mitochondria by mitophagy7. Mitochondrial morphology varies by tissue type8 and developmental stage9 and is regulated to allow cells to adapt to factors including energetic needs10,11 and stressors12. Standard morphometric features of mitochondria, such as the extent of network formation (interconnected vs. fragmented), perimeter, area, volume, length (aspect ratio), roundness, and degree of branching, can be measured and quantified by standard optical microscopy because the sizes of these features are greater than the diffraction limit of light (~200 nm)13.
Cristae architecture defines the internal structure of mitochondria (Figure 1B). The diversity of cristae morphologies can be broadly categorized as flat (lamellar or discoidal) or tubular-vesicular14. All cristae attach at the IBM through tubular or slot-like structures termed cristae junctions (CJs) that can serve to compartmentalize the IMS from the ICS and the IBM from the CM15. Cristae morphology is regulated by key protein complexes of the IMM, including (1) the mitochondrial contact site and cristae organizing system (MICOS) that resides at CJs and stabilizes IMM-OMM contacts16, (2) the optic atrophy 1 (OPA1) GTPase that regulates cristae remodeling17,18,19, and (3) F1FO ATP synthase that forms stabilizing oligomeric assemblies at cristae tips (CTs)20,21. In addition, the IMM is enriched in nonbilayer phospholipids phosphatidylethanolamine and cardiolipin that stabilize the highly curved IMM22. Cristae are also dynamic, demonstrating morphological changes under various conditions, such as different metabolic states23,24, with different respiratory substrates25, under starvation and oxidative stress26,27, with apoptosis28,29, and with aging30. Recently it has been shown that cristae could undergo major remodeling events on a timescale of seconds, underscoring their dynamic nature31. Several features of cristae can be quantified, including dimensions of structures within individual cristae (e.g., CJ width, crista length, and width) and parameters that relate individual crista to other structures (e.g., intra-cristae spacing and cristae incident angle relative to the OMM)32. These quantifiable cristae parameters show a direct correlation with function. For instance, the extent of mitochondrial ATP production is positively related to the abundance of cristae, quantified as cristae density or cristae number normalized to another feature (e.g., cristae per OMM area)33,34,35. Because IMM morphology is defined by nanoscale features, it comprises mitochondrial ultrastructure, which requires imaging techniques that provide resolution greater than the light diffraction limit. As described below, such techniques include electron microscopy and super-resolution microscopy (nanoscopy).
The neural and glial cells of the central nervous system (CNS) are particularly reliant on mitochondrial function. On average, the brain constitutes only 2% of the total body weight, but utilizes 25% of the total body glucose and accounts for 20% of body oxygen consumption, making it vulnerable to impairments in energy metabolism36. Progressive neurodegenerative diseases (NDs), including Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), Huntington's disease (HD), multiple sclerosis (MS), and Parkinson's disease (PD), are some of the most extensively studied pathologies to date, with research efforts ranging from understanding the molecular underpinnings of these diseases to seeking potential therapeutic prevention and interventions. NDs are associated with increased oxidative stress originating in part from reactive oxygen species (ROS) generated by the mitochondrial electron transport chain (ETC)37, as well as altered mitochondrial calcium handling38 and mitochondrial lipid metabolism39. These physiological alterations are accompanied by noted defects in mitochondrial morphology that are associated with AD40,41,42,43,44, ALS45,46, HD47,48,49, MS50, and PD51,52,53. These structural and functional defects can be coupled by complex cause-effect relationships. For example, given that cristae morphology stabilizes OXPHOS enzymes54, mitochondrial ROS are not only generated by the ETC, but they also act to damage the infrastructure in which the ETC resides, promoting a feed-forward ROS cycle that enhances susceptibility to oxidative damage. Furthermore, cristae disorganization has been shown to trigger processes such as mitochondrial DNA (mtDNA) release and inflammatory pathways connected to autoimmune, metabolic, and age-related disorders55. Therefore, analysis of mitochondrial structure is key to a full understanding of NDs and their molecular underpinnings.
Popular methods of viewing cristae, including transmission electron microscopy, electron tomography and cryo-electron tomography (cryo-ET), and X-ray tomography, in particular cryo-soft X-ray tomography, have revealed important findings and work with a variety of sample types56,57,58,59,60. Despite recent advancements toward better observation of organellar ultrastructure, these methods still come with the caveat of requiring sample fixation and, therefore, cannot capture real-time dynamics of cristae directly. Super-resolution fluorescence microscopy, particularly in the forms of structured illumination microscopy (SIM), stochastic optical reconstruction microscopy (STORM), photoactivated localization microscopy (PALM), expansion microscopy (ExM), and stimulated emission depletion (STED) microscopy, have become popular ways of viewing structures requiring resolution below the diffraction limit that constrains classical methods of optical microscopy. When ExM is used in conjunction with another super-resolution technique, the results are impressive, but the sample must be fixed and stained in a gel61. By comparison, SIM, PALM/STORM, and STED have all been successfully used with live samples, and new and promising dyes that generally stain the IMM provide a novel and easy approach for live imaging of mitochondria cristae dynamics62,63,64,65,66. Recent advancements in live dyes for STED imaging have improved dye brightness and photostability, and these dyes target the IMM at a higher degree of specificity than their predecessors. These developments allow the collection of long-term timelapse and z-stack experiments with super-resolution imaging, opening the door to better live cell analysis of mitochondrial ultrastructure and dynamics.
Herein, protocols for live cell imaging of undifferentiated and differentiated SH-SY5Y cells stained with the PKmito Orange (PKMO) dye using STED63 are provided. The SH-SY5Y cell line is a thrice subcloned derivative from the parental cell line, SK-N-SH, generated from a bone marrow biopsy of metastatic neuroblastoma67,68,69,70. This cell line is a commonly used in vitro model in ND research, particularly with diseases such as AD, HD, and PD, in which mitochondrial dysfunction is heavily implicated10,43,71,72,73. The ability to differentiate SH-SY5Y cells into cells with a neuron-like phenotype through manipulating culture media has proven a suitable model for neuroscience research without relying on primary neuronal cells10,74. In this protocol, retinoic acid (RA) was added to the cell culture medium to induce the differentiation of SH-SY5Y cells. RA is a vitamin A derivative and has been shown to regulate the cell cycle and promote the expression of transcription factors that regulate neuronal differentiation75. A protocol for culturing and live cell imaging of neurons isolated from the rat hippocampus is also provided. The hippocampus has been shown to be affected by mitochondrial degeneration and, along with the cortex, plays an important role in aging and ND76,77,78,79,80.
Protocol
1. Propagation and differentiation of SH-SY5Y cells
- Preparation of media for cell growth and maintenance
- Prepare complete, high-glucose Dulbecco's Modified Eagle's Medium (DMEM, 4.5 g/L D-glucose, 4 mM L-glutamine, 110 mg/L sodium pyruvate) supplemented with a final 1% (v/v) antibiotic-antimycotic (10,000 units/mL penicillin, 10,000 µg/mL streptomycin, and 25 µg/mL Amphotericin B), and varying amounts of fetal bovine serum (FBS) (see Table of Materials). FBS amounts in differentiation media vary between final 10%, 5%, or 2% (v/v) FBS.
- Cell maintenance
- Maintain the cells in DMEM supplemented with 10% (v/v) FBS and place them at 37 °C and 5% CO2, then seed in DMEM containing 5% (v/v) FBS for differentiation. Frozen cell stocks were stored in FBS with 10% (v/v) dimethyl sulfoxide (DMSO) at 1-2 x 107 cells/mL.
- Preparation of retinoic acid (RA)
- Dissolve 7.51 mg all-trans-RA (see Table of Materials) in 5 mL of freshly prepared 95% ethanol to obtain 5 mM stock solution. Verify concentration with absorbance at 350 nm (ɛ = 44,300 M-1 cm-1), obtained from the product information sheet from the manufacturer's protocol81, using a dilution of the stock solution at 5 µM in ethanol. Store 5 mM stock protected from light at 4 °C for up to 6 weeks.
- Preparation of poly-D-lysine for coverslip coating
NOTE: The poly-D-lysine (PDL) product protocol, found under the Documents & Downloads section of vendor site82, provides information for coating a variety of culture vessels.- This protocol includes volumes based on a 2-well chambered vessel with an area of 4 cm2 per well with sterile #1.5 borosilicate coverglass bottoms (see Table of Materials). Dilute the stock solution of PDL twofold to 50 µg/mL with Dulbecco's PBS (DPBS; no calcium, no magnesium).
NOTE: #1.5 or #1.5H coverglass are both acceptable thicknesses, which are essential for image quality. Other thicknesses will induce spherical aberration and should be avoided.
- This protocol includes volumes based on a 2-well chambered vessel with an area of 4 cm2 per well with sterile #1.5 borosilicate coverglass bottoms (see Table of Materials). Dilute the stock solution of PDL twofold to 50 µg/mL with Dulbecco's PBS (DPBS; no calcium, no magnesium).
- Coverslip coating with PDL
NOTE: Coverslips can be exposed to ultraviolet (UV) light for 10-15 min in a biosafety cabinet for further sterilization.- Apply 1.2 mL of 50 µg/mL PDL solution to each well of sterile chambered coverslips in a cell culture cabinet and incubate at room temperature for 1 h. Remove the PDL solution and rinse thrice with 3.6 mL distilled water. After completing the final wash, allow the coated chamber to dry for 2 h exposed to air before rinsing and using immediately or storing with an air-tight container at 4 °C for up to 2 weeks.
NOTE: Rinse the coverslips thoroughly as excess PDL can be toxic to cells.
- Apply 1.2 mL of 50 µg/mL PDL solution to each well of sterile chambered coverslips in a cell culture cabinet and incubate at room temperature for 1 h. Remove the PDL solution and rinse thrice with 3.6 mL distilled water. After completing the final wash, allow the coated chamber to dry for 2 h exposed to air before rinsing and using immediately or storing with an air-tight container at 4 °C for up to 2 weeks.
- Differentiation of SH-SY5Y cells with RA
NOTE: Do not use cells above passage 15. Cells are passaged at 80%-90% confluency. Differentiation procedures differ but follow similar steps. Additional differentiation from neuroblastomas to mature neurons is obtained with further treatment with brain-derived neurotrophic factor (BDNF)68,83,84,85, but was not performed in this protocol.
OPTIONAL: Establish cells for at least 24 h prior to seeding on coverglass. To prepare cells from frozen stocks, rapidly thaw 1 mL frozen vial of cells and add to 9 mL prewarmed media supplemented with 10% FBS, then spin down at 350 x g for 10 min (at room temperature) and discard supernatant to remove DMSO. Resuspend the cell pellet in 5 mL of prewarmed media and seed cells in a T-25 flask. Once cells reach 80%-90% confluency, passage cells by counting and seeding them for differentiation when applicable.- Day 0: Seed cells.
- Seed the cells onto a chambered coverglass from frozen stocks or a working flask. Use a seeding density of 1.5 x 104 cells/cm2.
NOTE: A single well in a standard 2-well chambered coverglass with 4 cm2 of culture area will require 6.0 x 104 cells. Cells that will remain undifferentiated should be seeded with DMEM supplemented with 10% (v/v) FBS, and cells that will be differentiated should be seeded with DMEM supplemented with 5% (v/v) FBS.
- Seed the cells onto a chambered coverglass from frozen stocks or a working flask. Use a seeding density of 1.5 x 104 cells/cm2.
- Day 1: Start RA differentiation treatment.
- Prepare DMEM supplemented with 5% (v/v) FBS, 1% (v/v) antibiotic-antimycotic, and a final concentration of 10 µM RA or ethanol of the same additive volume to serve as the vehicle control for this differentiation procedure. Remove the media in the chambered coverglass used for seeding, rinse with 1x PBS, and add the new DMEM to the wells.
- Day 3: Replace media with fresh RA- or ethanol-containing media.
- Remove media from Day 1 and replace with fresh media supplemented with 2% (v/v) FBS, 1% (v/v) antibiotic-antimycotic, and either 10 µM RA or 95% ethanol of the same additive volume to serve as the vehicle control for this differentiation procedure. Remove the media in the chambered coverglass used for seeding, rinse with 1x PBS, and add new media to the wells.
- Day 6: Perform the imaging of the cells.
NOTE: Cell differentiation times vary by protocol, but six days of exposure to RA is sufficient to induce a neuron-like phenotype in SH-SY5Y cells86.- Perform live imaging, with details in Sections 3 and 4 (Figure 2).
- Day 0: Seed cells.
2. Primary rat hippocampal neuron culture
- Materials preparation for rat hippocampal neuron isolation.
- Prepare fresh supplemented DMEM.
- Filter-sterilize a mixture of DMEM (high glucose, no sodium pyruvate) supplemented with 10% (v/v) heat-inactivated FBS, 1% (v/v) sodium pyruvate solution, and 1% (v/v) penicillin-streptomycin (10,000 U/mL). Store for up to 2 weeks at 4 °C.
- Prepare fresh supplemented neuron growth media.
- Filter-sterilize a mixture of neuron growth media supplemented with 2% (v/v) B27 supplement, 0.25% (v/v) glutamine supplement, and 1% (v/v) penicillin-streptomycin (see Table of Materials). Store for up to 2 weeks at 4 °C.
- Prepare fresh supplemented DMEM.
- Prepare primary hippocampal neuron culture.
- Prepare primary hippocampal neuron culture following previously published work87 and from the product protocol on the manufacturer's site from which the dissected E18 rat hippocampus is obtained88 (see Table of Materials). This protocol results in a mostly neuronal population with <2% astrocytes.
NOTE: The hibernate media with which this tissue is shipped will be used for future steps in this protocol. Do not discard it. - Prepare materials and media for tissue dissociation.
- Flame a Pasteur pipette to decrease the aperture diameter and store it in foil to prevent contamination until use. Prewarm the prepared DMEM, 1X Hank's Buffered Salt Solution (HBSS), and neuron growth media to 37 °C. Add 2 flakes of DNAase with sterile tweezers to a 15 mL conical tube.
- Perform tissue dissociation.
- Remove as much of the hibernate media the dissected E18 rat hippocampus is stored in as possible before placing the tissue into the 15 mL conical tube containing DNAase and incubating briefly at 37 °C. Add 900 µL of 1X HBSS followed by 100 µL of 0.5% trypsin. Incubate the tissue at 37 °C for 15 min.
NOTE: PDL coated plates can be removed from storage and placed in an incubator until use during this incubation time.
- Remove as much of the hibernate media the dissected E18 rat hippocampus is stored in as possible before placing the tissue into the 15 mL conical tube containing DNAase and incubating briefly at 37 °C. Add 900 µL of 1X HBSS followed by 100 µL of 0.5% trypsin. Incubate the tissue at 37 °C for 15 min.
- Perform tissue homogenization and cell counting.
- Following incubation with trypsin, remove the media and add 1 mL prewarmed hibernate media-DNAase from the previous step to the tissue and homogenize with the Pasteur pipette. The media will look opaque and then gradually clear as homogenization continues.
- Add dissociated neurons to a new tube with 4 mL prewarmed DMEM and then count the cells using a cell counter.
- Perform cell plating and growth of primary cells.
- Plate cells at a density of approximately 1.65 x 104 cells/cm2 in DMEM. For a 2-well chambered coverglass (4 cm2), seed 65,000 – 70,000 cells per well with 2 mL DMEM. Incubate cells at 37 °C and 5% CO2 for 2 h before checking for adherence. Once cells begin to adhere, remove 1 mL of media and replace it with the same volume of hibernate media, then gently agitate to mix. Once the media is mixed, repeat this process and remove half of the media present and replace with the same volume of neuron growth media, then gently mix.
NOTE: The day of plating is considered day in vitro (DIV) 0, and cells are ready to image at DIV 7 (Figure 2).
- Plate cells at a density of approximately 1.65 x 104 cells/cm2 in DMEM. For a 2-well chambered coverglass (4 cm2), seed 65,000 – 70,000 cells per well with 2 mL DMEM. Incubate cells at 37 °C and 5% CO2 for 2 h before checking for adherence. Once cells begin to adhere, remove 1 mL of media and replace it with the same volume of hibernate media, then gently agitate to mix. Once the media is mixed, repeat this process and remove half of the media present and replace with the same volume of neuron growth media, then gently mix.
- Prepare primary hippocampal neuron culture following previously published work87 and from the product protocol on the manufacturer's site from which the dissected E18 rat hippocampus is obtained88 (see Table of Materials). This protocol results in a mostly neuronal population with <2% astrocytes.
3. Preparation of cells for live cell imaging
NOTE: Cell types and origin (i.e., cultured and primary cells) can differ in staining requirements; see published reports for more details62,63.
- Preparation of PKmito Orange
NOTE: Other dyes that generally stain the IMM have been reported64,65,66 and are commercially available. PKMO is the only one used in this protocol.- Resuspend powder PKMO (see Table of Materials) in DMSO as per the manufacturer's instructions89. Aspirate the media from cells and wash in prewarmed phenol-red free media. Prepare a stock of PKMO in prewarmed, phenol red-free DMEM supplemented with 2% (v/v) FBS or 10% (v/v) depending on differentiation state, HEPES to a final concentration of 20 mM, and 1% (v/v) antibiotic-antimycotic before staining the cells following the manufacturer's instructions. This formulation, without PKMO, is the live cell imaging media.
- Cell staining with PKMO
- Incubate the cells with the dye at 37 °C and 5% CO2, for 30 min. Wash cells thrice with live cell imaging media, and for the final wash incubate for 30 min at 37 °C, 5% CO2.
- Add fresh, prewarmed live cell imaging media. The cells are now ready for imaging.
NOTE: Acute treatments (e.g., drugs and/or stressors), when used, are added before live imaging; see the Discussion section and Supplementary Figure 1.
4. Imaging live cells by STED microscopy
NOTE: This protocol uses a STED system built around an inverted microscope, with the system specified in the Table of Materials. This system is equipped with pulsed excitation lasers (561 nm laser with nominal power ~300 µW) and a pulsed 775 nm STED depletion laser (nominal power 1.2 W), a continuously adjustable galvano scanner, and a 615/20 nm filter-based avalanche photodiode detector (APD). A 100x/1.40 oil immersion lens for STED is used here. Lightbox software is used for image acquisition. All details provided are related directly to this software and system setup.
- General guidelines and steps for imaging
- Use a stage-top incubator or environmental chamber to maintain cell viability, but short-term room-temperature experiments are also acceptable. These steps are specific to the STED setup described above.
- Select the laser and filter sets for imaging.
- Use parameters for an orange dye by selecting the dye(s) used in the staining from the Dye List, or the one with spectral properties closest to the dye(s) used. Make these active by double-clicking or dragging them to the sample list, where it says “Drag dye(s) here.”
- Select a region to image.
- In an overview, create a region of interest (ROI) around a mitochondrion of interest by selecting the rectangular ROI button and clicking and dragging to shape the region. The ROI can be resized and rotated using the ROI corners or curved edges that appear while hovering the mouse over a corner.
NOTE: A summary of the suggested imaging parameters can be found in Table 1. These parameters were empirically adjusted using those previously reported for this STED setup and dye combination63.
- In an overview, create a region of interest (ROI) around a mitochondrion of interest by selecting the rectangular ROI button and clicking and dragging to shape the region. The ROI can be resized and rotated using the ROI corners or curved edges that appear while hovering the mouse over a corner.
- Set up gating.
- Next to the General menu, select the Gating menu or click and hold to add the menu to the view. It is recommended that STED detector gating be adjusted to 1-1.05 to 7.8-7.85 ns, as presented here. Gating times can vary and be as short as 1-1.05 to 7-7.05 ns.
- Adjust the intensity appropriately according to the sample.
NOTE: Generally, the excitation power used for STED is 2-3 times the power used for confocal, so a sample requiring 5% excitation laser power for confocal can use 10%-15% excitation laser power during STED acquisition.- Set the excitation laser for STED to 15%-20% and the STED depletion laser to 20%-25% with 10 line accumulations. Use a pixel dwell time of 4 µs with a pixel size of 20-25 nm. The pinhole was kept at 1.0 AU for cultured cells and 0.7 AU for primary rat hippocampal cells in order to improve optical sectioning in the more densely packed mitochondria.
NOTE: Confocal and STED images can both be acquired for side-by-side comparison (Figure 3A, B, Figure 4A) or only STED can be acquired.
- Set the excitation laser for STED to 15%-20% and the STED depletion laser to 20%-25% with 10 line accumulations. Use a pixel dwell time of 4 µs with a pixel size of 20-25 nm. The pinhole was kept at 1.0 AU for cultured cells and 0.7 AU for primary rat hippocampal cells in order to improve optical sectioning in the more densely packed mitochondria.
- Additional information for time series/z-series
- Select the time series.
- Select the Time drop-down menu. Define the number of iterations (5 used here) and time interval (25 or 30 s used here) desired for a timelapse (Figure 3C, D, Figure 4B).
NOTE: If the interval is shorter than the acquisition time, the iterations will continue without any delay. When performing a timelapse, engaging a perfect focus unit or similar focus tracking is highly recommended to avoid potential drift.
- Select the Time drop-down menu. Define the number of iterations (5 used here) and time interval (25 or 30 s used here) desired for a timelapse (Figure 3C, D, Figure 4B).
- Set the volume range.
- Set the z-volume range as desired by enabling the Volume option and adjusting the two ends of the range. Step sizes used in this protocol for 2D imaging were 150-200 nm. Recommended step size with respect to Nyquist sampling required for deconvolution of raw STED can be calculated with online tools90.
NOTE: The STED depletion laser power and the number of planes imaged can increase dye photobleaching and light exposure to the cell to harmful levels. Check for signs of phototoxicity and photodamage after acquisition.
- Set the z-volume range as desired by enabling the Volume option and adjusting the two ends of the range. Step sizes used in this protocol for 2D imaging were 150-200 nm. Recommended step size with respect to Nyquist sampling required for deconvolution of raw STED can be calculated with online tools90.
- Select the time series.
5. Processing and analytical tools for mitochondrial ultrastructure
NOTE: Image processing (i.e., deconvolution) is optional but typically used when making and analyzing STED images for publication. Deconvolution to improve contrast and reduce noise is highly suggested for optimal segmentation of individual cristae, as described below (Figure 2).
- STED image deconvolution
NOTE: The software used for deconvolution in this protocol is provided in the Table of Materials.- Set the microscopic parameters of the image.
NOTE: Ensure that microscope metadata is entered correctly in the image Microscopic Parameters. This includes mounting medium refractive index; immersion medium; pixel size; excitation, emission, and depletion wavelengths; and any other relevant information. Templates with these parameters can be saved for reuse. - Deconvolute raw STED images with the software algorithm.
- Access to automated deconvolution algorithms in deconvolution software allows hands-off deconvolution image processing. Select the Express button and set the deconvolution type to Fast, Standard, Aggressive, or Conservative for varying degrees of deconvolution power. Representative images using Express Deconvolution with Aggressive settings are shown (Figure 3, Figure 4, and Figure 5).
- Save the images from the deconvolution software in ICS2 format.
- For manual deconvolution, perform the following steps.
- Briefly, when performing manual deconvolution, save the deconvolution wizard templates for consistency and have the option to load a template upon starting the wizard. Use measured point spread function (PSF) information if generated with acquisition setup and parameters.
- Crop raw STED image, if needed, before having the deconvolution software auto-stabilize the image. Add-ons to deconvolution software packages allow specific compensation of possible imaging artifacts such as thermal drift and chromatic aberrations.
- Next, generate a logarithmic histogram to manually or automatically perform background subtraction. Select the classic maximum likelihood estimation (CMLE) deconvolution algorithm.
NOTE: For deconvolution, the key values to adjust are the signal-to-noise ratio threshold, the number of iterations, and the quality threshold. These values can be adjusted, and the deconvolution can be previewed to determine optimal settings.
- Set the microscopic parameters of the image.
- Segmentation and particle analysis
NOTE: This protocol uses FIJI (Is Just ImageJ), an open-source software (see Table of Materials), for segmentation and analysis. Other comparable software, including CellProfiler, Icy, Ilastik, and QuPath, is available for these purposes.- Prepare images for segmentation.
- Open the .obf raw STED images or .ics2 files from the deconvolution software by going to File → Open or clicking and dragging the files to the ImageJ toolbar. From here, any processing that makes images easier to segment can be performed before segmentation.
- To keep changes consistent, record functions using Plugins → Macros → Record and copy and paste key commands into a new macro, from Plugins → New → Macro. Ensure to select the image to process before running the macro.
NOTE: Commonly acceptable alterations for quantitation of size and shape include cropping, projecting a z-stack, and background subtraction with smoothing disabled. Alterations should be performed consistently among images within a data set and reported.
- Adjust Trainable Weka Segmentation Settings
NOTE: Additional details with step-by-step instructions for using the semi-automated segmentation tool and downstream analyses for mitochondria have been published91.- Open the deconvolved STED images in the Trainable Weka Segmentation (TWS)92 plugin, located under Plugins → Segmentation → Trainable Weka Segmentation. In the segmentation Settings, select the Gaussian blur, membrane projections, and Sobel filter features. The membrane thickness default is 1, and the membrane patch size default is 19.
- Label either class 1 or 2 as the "Cristae" and the other as the "Background" (Figure 2). Models can also be saved with the Save classifier button. Select the Load classifier to reuse these settings for other images.
- Perform TWS class traces.
- Use the line tool or other shapes to highlight some of the cristae or background. At least some background selections should include spaces between the cristae. Draw a line over the structure to assign to either class, then select the Add to button on the right-hand side for either cristae or background. Double-click on a trace to remove the structure from that label.
- Perform TWS classification training.
- Select the Train classifier button on the left-hand side to generate a map based on the information provided to the plugin. The overlay of segmented classes can be toggled on and off with the Toggle Overlay button, and the opacity of the overlay can be adjusted in Settings. The classifier can be retrained with additional labels. Once satisfied, select the Get Probability button.
- Measure the particles.
- Using the cristae probability map, threshold the image to generate a mask and then go to Analyze → Analyze Particles. Generally, the default threshold type can be used and the range adjusted to ensure the entire cristae are accounted for. The measurements provided by analyzing particles can be adjusted by Analyze → Set Measurements.
NOTE: Examples of size and shape parameters such as area, perimeter, circularity and aspect ratio of the cristae are measured and displayed based on the selected measurements (Figure 2, Figure 5A, Supplementary Table 1). - OPTIONAL: Select the raw STED image with the same dimensions as the deconvolved image and apply the ROIs from the manager, then select Measure in the ROI manager to obtain intensity values.
- Using the cristae probability map, threshold the image to generate a mask and then go to Analyze → Analyze Particles. Generally, the default threshold type can be used and the range adjusted to ensure the entire cristae are accounted for. The measurements provided by analyzing particles can be adjusted by Analyze → Set Measurements.
- Obtain line plots.
- Line plots were generated from the deconvolved STED images. Draw a multi-point line, adjust the line thickness to several pixels wide to average out noise, and spline the line to fit the mitochondria (Figure 2, Figure 5B). The resulting line plot generated is used to measure peak-to-peak distances to report the periodicity and distribution of cristae over a certain range.
NOTE: Relatedly, cristae density can be reported as the count of independent cristae within a given area as determined by measuring the outer part of the mitochondria. The mitochondrial area can be determined by applying a filter to the deconvolved or raw STED image to generate a mask. Ensure that the mask accurately fits the outline of the mitochondria before measuring the area.
- Line plots were generated from the deconvolved STED images. Draw a multi-point line, adjust the line thickness to several pixels wide to average out noise, and spline the line to fit the mitochondria (Figure 2, Figure 5B). The resulting line plot generated is used to measure peak-to-peak distances to report the periodicity and distribution of cristae over a certain range.
- Prepare images for segmentation.
Representative Results
This protocol describes cell growth conditions for cultured and primary cells with a focus on live cell STED imaging and subsequent analyses of mitochondrial cristae. Projections made with ImageJ of mitochondria from undifferentiated SH-SY5Y (Figure 3A) and RA-differentiated SH-SY5Y (Figure 3B) cells can be collected as z-stacks with traditional confocal and STED, and the raw STED images can then be deconvolved. Timelapse imaging can also be performed and subsequently deconvolved (Figure 3C,D). Using slightly different imaging parameters for primary rat hippocampal neurons (Table 1), confocal and raw STED images can be acquired as z-stacks, and the raw STED images can be deconvolved (Figure 4A). Timelapse imaging of mitochondria from primary neurons is also possible (Figure 4B). In general, the time-lapse images should be able to show mitochondrial dynamic events.
When raw STED and deconvolved STED z-stack projections from the samples used for segmentation appear consistent, quantitative measurements are performed. The TWS plugin uses the deconvolved STED image to segment to make a probability mask, which is then used to create a binary mask of the cristae to obtain size and shape parameters (Figure 5A). The regions from this mask are saved in the ROI manager and can be applied to the raw STED image if desired to measure differences in relative intensity. The deconvolved STED projections can also be used to determine the cristae periodicity and density in a given area (Figure 5B).
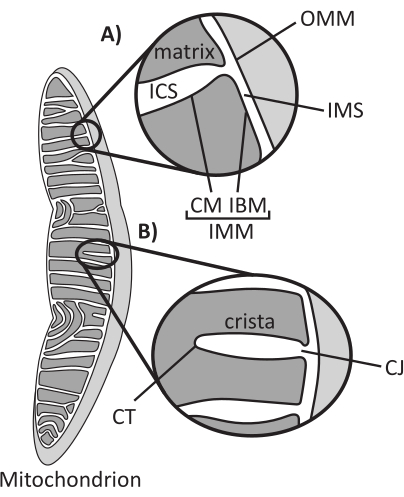
Figure 1: Mitochondrial morphology. Mitochondria have a two-membrane system that defines different subcompartments (A). Cristae are infolds of the inner membrane with defined features (B). Abbreviations: OMM, outer mitochondrial membrane; ICS, intracristal space; IMS, intermembrane space; CM, cristae membrane; IBM, inner boundary membrane; IMM, inner mitochondrial membrane; CT, cristae tip; CJ, cristae junction. Please click here to view a larger version of this figure.
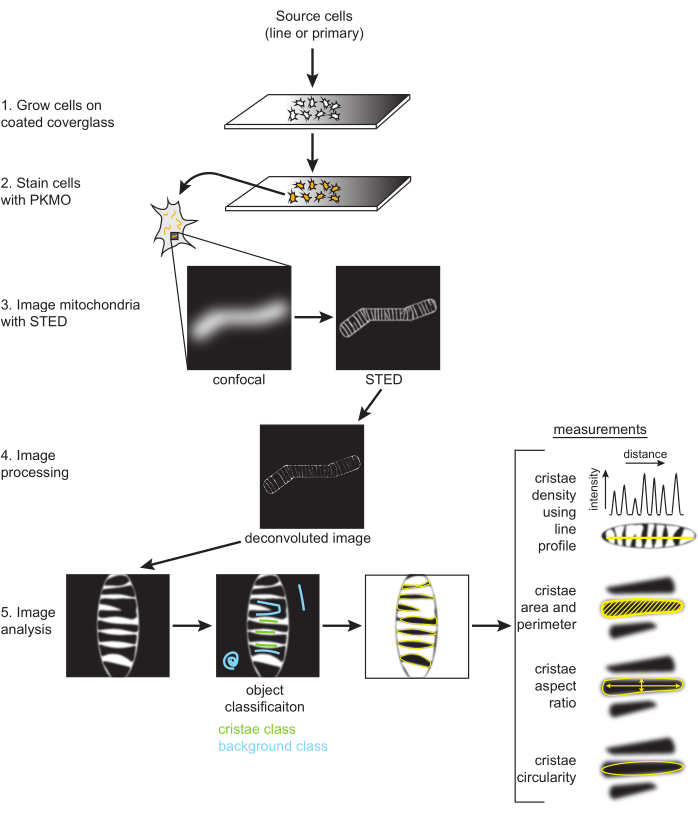
Figure 2: Schematic of workflow. SH-SY5Y cells or primary rat hippocampal neurons are grown on a PDL-coated coverglass. SH-SY5Y cells are grown in parallel to remain undifferentiated or subjected to RA differentiation over the course of six days. Primary rat hippocampal neurons were grown on a PDL-coated coverglass after being isolated from hippocampal sections for seven days. Once ready to be imaged, cells were stained with PKMO and imaged with STED. Raw STED images are then deconvolved, and the deconvolved images are processed in FIJI to obtain size and shape measurements, such as cristae density, area, perimeter, circularity, and aspect ratio. Please click here to view a larger version of this figure.
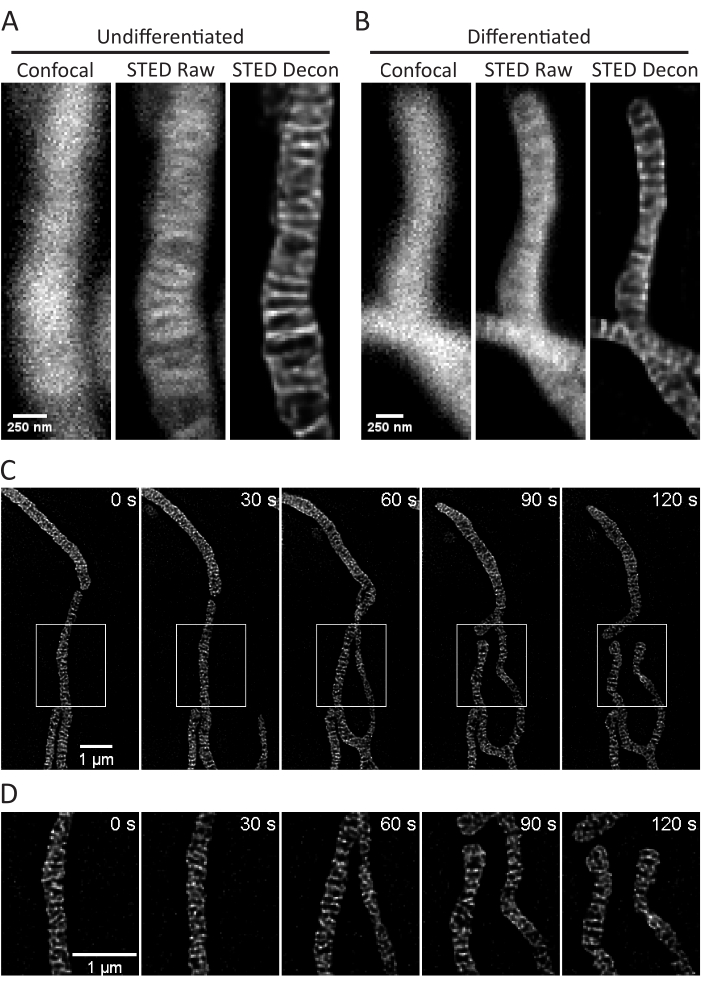
Figure 3: Imaging of mitochondria in SH-SY5Y cells. Representative confocal (left), raw STED (center), and Huygens deconvolved STED image z-stack projections (right) of mitochondria from non-differentiated (A) and RA-differentiated (B) SH-SY5Y cells with PKMO staining are shown. A timelapse with 30 s intervals and 5 iterations of RA-differentiated SH-SY5Y cells is shown (C) with selected regions (white boxes) expanded upon (D) using scaled images of those regions without interpolation. Scale bars: A,B, 250 nm; C,D, 1 µm. Please click here to view a larger version of this figure.
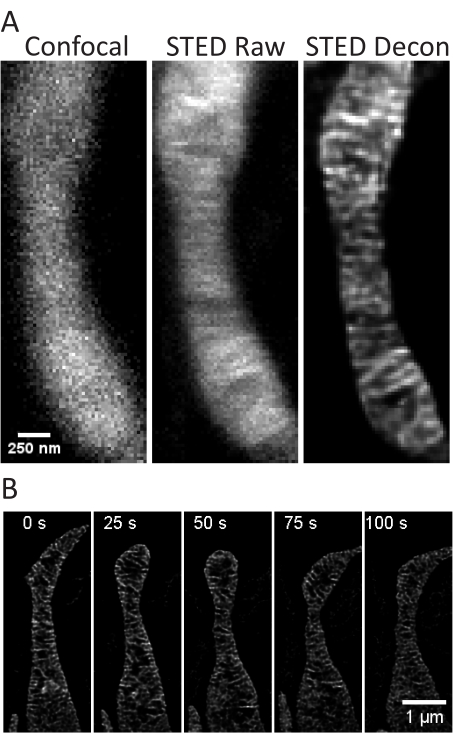
Figure 4: Imaging of mitochondria in primary rat hippocampal neurons. Representative confocal (left), raw STED (center), and Huygens deconvolved STED (right) image z-stack projections of mitochondria from primary rat hippocampal neurons are shown (A). A timelapse with 25 s intervals and 5 iterations of mitochondria in these neurons is shown (B). Scale bars: A, 250 nm; B, 1 µm. Please click here to view a larger version of this figure.
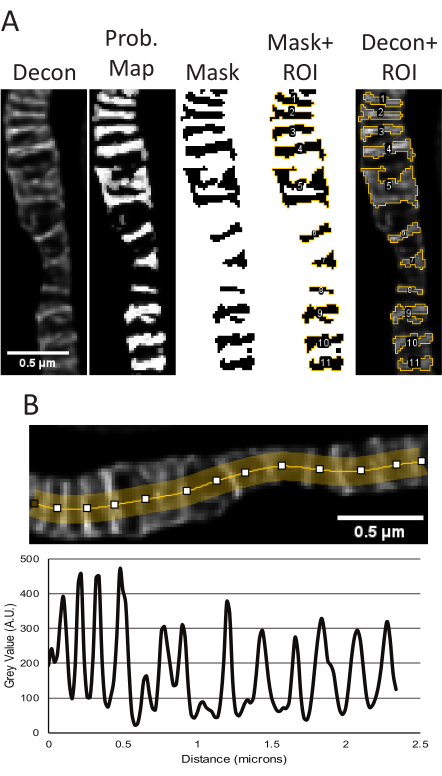
Figure 5: Processing of deconvolved STED images in ImageJ. Representative use of the Trainable Weka Segmentation plugin to measure cristae size and shape is shown (A). From left to right, the following images are shown: the deconvolved STED image, the probability map based on segmentation from the TWS plugin, the mask from thresholding in FIJI using the probability map as input, the mask with the ROIs outlined, and the ROIs overlayed onto the original deconvolved STED image. The resulting area, perimeter, circularity and aspect ratio measurements corresponding to these objects can be found in Supplementary Table 1. A line plot using the deconvolved STED image to measure peak-to-peak distances as a readout for cristae density is shown (B). Scale bars: 0.5 µm. Please click here to view a larger version of this figure.
| Pixel size (nm) | Dwell time (µs) | Line acc. | 561 nm excitation during STED acquisition (%) | 775 nm STED depletion power (%) | Step size (nm) | Pinhole (AU) | Timelapse interval (s) | Timelapse iterations | |
| Undiffer-entiated SH-SY5Y | 20 | 4 | 10 | 15 | 20 | 200 | 1.0 | 30 | 5 |
| RA-Differe-ntiated SH-SY5Y | 20-25 | 4 | 10-12 | 15 | 20-22 | 150-200 | 1.0 | 30 | 5 |
| Primary Neurons | 20-25 | 4 | 10 | 10 | 25 | 200 | 0.7 | 30 | 5 |
| NOTE: Pixel size can vary based on imaging requirements and intent to deconvolve images. Proper sampling is required for deconvolution. Pixel sizes for raw STED images without deconvolution can go up to 30 nm. | |||||||||
Table 1: Summary of STED acquisition parameters. The settings used for 2D STED imaging for each cell type, undifferentiated SH-SY5Y, RA-differentiated SH-SY5Y, and primary rat hippocampal neurons, are displayed. For all time-lapses, 5 iterations were taken with varying intervals based on ROI size.
Supplementary Figure 1: Imaging of SH-SY5Y cells with amyloid-β (Aβ) addition. Representative confocal (left), raw STED (center), and deconvolved STED (right) images of RA-differentiated SH-SY5Y cells with PKMO stain (top) and Aβ-HiLyte647 (bottom) are shown (A). Merged z-stack projections of raw PKMO STED (green) with raw Aβ STED (magenta) (B) or deconvolved PKMO STED (green) with deconvolved Aβ STED (magenta) (C) are shown. Scale bars: 0.5 µm. Please click here to download this File.
Supplementary Table 1: Size and shape measurements of segmented cristae. The size and shape measurements of area (µm2), perimeter (µm), circularity, and aspect ratio, corresponding to the objects outlined in Figure 5A from segmented mitochondria, are shown. Please click here to download this File.
Supplementary Table 2: Summary of acquisition parameters with amyloid-β samples. The settings used for 2D STED imaging of PKMO and Aβ-HiLyte647 in undifferentiated and RA-differentiated SH-SY5Y cells are displayed. Confocal of Aβ-HiLyte647 may be used alone as there is no specific structure to resolve; STED images of Aβ-HiLyte647 are shown here for smaller particle sizes. Please click here to download this File.
Supplementary File 1: Amyloid-β treatment protocol. Please click here to download this File.
Discussion
This protocol presents the use of human neuroblastoma cell line SH-SY5Y and primary rat hippocampal neurons with the novel IMM-targeting PKMO dye for live cell STED imaging. Due to the novelty of PKMO, there is currently little published using this dye for live STED imaging. Using these cell types for STED imaging poses challenges, specifically because neuronal cells have narrower mitochondria. One limitation of this protocol is the PKMO dye used, as it can be toxic to cells. Different cells and cell lines respond differently to the dye, thus, adjustments to dye concentration and incubation time to optimize results for strong signal without harming cells may be required. A suggested solution is to lower the concentration and increase the staining time63; however, this may result in poorer staining without increasing cell viability.
Similarly to PKMO, the commercial dye Live Orange mito (Table of Materials) also exhibits some cell toxicity. This dye was used for a variety of cultured cells but was unable to exhibit comparable staining in RA-differentiated SH-SY5Y cells successfully with the same parameters as their undifferentiated counterparts (our unpublished observations). However, amenable staining protocols may be optimized for this probe and chosen cell types. With this dye, detector gating times of 1-1.05 to 7-7.05 ns were used, with all other parameters in Table 1 remaining the same. Generally, staining cells with 200-250 nM Live Orange mito for 45 min yielded comparable results as the PKMO results shown. Higher concentration staining for less time or lower concentration staining for the same amount of time or slightly longer can yield different results and may be favorable to other cell types or growth conditions.
Imaging primary rat hippocampal neurons differs from immortalized cells due to the nature of the axon and dendrite projections as well as mitochondrial distribution at the time of imaging. One difficulty in this part of the protocol is that seeding density determines whether the primary cultures will be able to adhere and grow healthily, and at higher densities, the projections tend to overgrow by DIV 10. Therefore, the mitochondria imaged from these primary neurons will likely come from the cell body and not the projections; however, successful growth from a lower starting cell density yields better imaging results at later growth times. The key is to ensure low background and out-of-focus light to have the best contrast for STED. To address concerns regarding cell population, culturing primary hippocampal cells in B27-supplemented neuron growth media prevents the growth of glial cells, and the source reports that <5% of cells are astrocytes and the absence of NbActiv1 supplement in the growth media reduces the number of astrocytes in cultures to <2%87. For both cultured SH-SY5Y cells and primary rat hippocampal neurons, the PDL coating used for growth contributes to background haze in images. Sufficient signal-to-noise is accomplished with the settings reported in (Table 1) and deconvolution removes most of the background observed.
In addition to the imaging covered here, it is also possible to add treatments or stress to cells before or during imaging. For example, adding tert-butyl hydrogen peroxide (tBHP) induces oxidative stress, and it is possible to monitor changes in mitochondria over time after addition. The addition of amyloid-β (Aβ) with a fluorescent tag allows monitoring of the distribution of this peptide in relation to mitochondria as well as the mitochondrial structure over time. Mitochondrial health has been heavily implicated in AD and is widely supported to play a role in Aβ toxicity43,71,72. Notably, the differentiation status of SH-SY5Y cells affects Aβ protein precursor (AβPP) localization85, and experiments using AβPP should be carefully constructed.
As an example of how this protocol can be adapted, it is shown that the fluorescent variant Aβ(1-42)-HiLyte 647 can be added to PKMO-stained cells 15 min before imaging (Supplementary Figure 1). The imaging parameters are similar (Supplementary Table 2), with the main difference being that a smaller pinhole is needed when imaging narrower mitochondria. Imaging Aβ-HiLyte647 with STED requires less overall excitation (6%-8%) and STED depletion (10%-12%) laser power and fewer accumulations (six). Detector gating is also extended from 0.1 to 10 ns. Although STED resolution of Aβ is not necessary, the overall signal-to-noise ratio and Aβ particle size of raw STED were better than those of the confocal images, and subsequent deconvolution can also be performed. Collecting STED images and deconvolving raw STED z-stack projections of Aβ appears particularly useful when merging with raw STED or deconvolved STED images of the PKMO stain (Supplementary Figure 1B,C). Both channels were collected in a single frame step. Measurements of time-dependent localization, similar to those listed in Figure 2 and shown in Figure 5, where applicable, and cristae architecture differences can be obtained following stress treatment or other additions.
Other possible methods for dual-labeling in live cell STED of mitochondria not reported here but have been reported by others include the use of SNAP-tagged proteins93, Halo-tagged proteins, and the use of other cell-permeable dyes with generic targets, such as mtDNA63. Notably, the labeling strategy of SNAP and Halo tagging influences the resulting fluorescence signal intensity and longevity when imaging94. Additionally, while this protocol presents several examples of analyses that can be applied to segmented mitochondria, there are many other analyses that software packages can perform on these images.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Primary rat hippocampal neurons were supplied by Dr. George Lykotrafitis and Shiju Gu of the Biomedical Engineering Department at the University of Connecticut (Storrs, CT, USA). The Abberior STED instrument housed in the Advanced Light Microscopy Facility in the Center for Open Research Resources and Equipment was acquired with NIH grant S10OD023618 awarded to Christopher O'Connell. This research was funded by NIH grant R01AG065879 awarded to Nathan N. Alder.
Materials
| 0.05% Trypsin-EDTA | Gibco | 25300054 | |
| 0.4% Trypan blue | Invitrogen | T10282 | |
| 0.5% Trypsin-EDTA, no phenol red | Gibco | 15400054 | |
| 100 X antibiotic-antimycotic | Gibco | 15240062 | |
| 100 X/1.40 UPlanSApo oil immersion lens | Olympus | Equipped in Olympus IX83 microscope for STED setup described in Section 4 | |
| All-trans-retinoic acid | Sigma | R2625 | |
| Amyloid-β (1-42, HiLyte Fluor647, 0.1 mg) | AnaSpec | AS-64161 | Other fluorescent conjugates available |
| B27 supplement (50 X), serum free | Gibco | 17504044 | |
| Cell Counter (Countess II FL) | Life Technologies | AMQAF1000 | |
| Centrifuge | Eppendorf | 5804-R | |
| Counter slides | Invitrogen | C10283 | |
| Conical tubes (15 mL) | Thermo Fisher Scientific | 339650 | |
| Cuvettes (Quartz Cells) | Starna Cells, Inc. | 9-Q-10 | Used with Spectrometer as described in Section 1.3 |
| DMEM (high glucose with sodium pyruvate) | Gibco | 11995073 | Used for SH-SY5Y cell materials as described in Section 1 |
| DMEM (high glucose no sodium pyruvate) | Gibco | 11965092 | Used for primary cell materials as described in Section 2 |
| DMEM (phenol red-free) | Gibco | 31053028 | Used for imaging as described in Section 3 |
| DMSO | Sigma | D8418 | |
| DNAase I from bovine pancreas | Sigma | DN25 | Used for primary cell materials as described in Section 2.2.1 and 2.2.2 |
| DPBS (no calcium, no magnesium) | Gibco | 14190144 | |
| E18 Rat Hippocampus | Transnetyx Tissue | SDEHP | |
| Ethanol (200 proof) | Fisher Bioreagents | BP28184 | |
| Fetal bovine serum (FBS), not heat-inactivated | Gibco | 26140079 | For cultured cells, in Section 1 |
| Fetal bovine serum (heat inactivated) | Gibco | 10082147 | For primary cell culture, Section 2 |
| Filter sterilization unit (0.1 µm, 500 mL) | Thermo Fisher Scientific | 5660010 | |
| FIJI (Is Just ImageJ) and Trainable Weka Segmentation (TWS) plug-in | — | — | Free, open-source image analysis software that includes plug-ins including Trainable Weka Segmentation described in Section 5; TWS plug-in from ref. 90 of the main text |
| GlutaMAX supplement (100 X) | Gibco | 35050061 | Glutamine supplement used for primary cell materials described in Section 2.1.2 |
| Hausser Scientific bright-Line and Hy-Lite Counting Chambers | Hausser Scientific | 267110 | |
| HBSS (no calcium, no magnesium) | Gibco | 14170120 | Used for primary cell materials described in Section 2.2.1 and 2.2.2 |
| HEPES | Gibco | 15630080 | |
| Huygens Professional deconvolution software (V. 20.10) | Scientific Volume Imaging (SVI) | — | The deconvolution software used in this protocol and described in Section 5 |
| IX83 inverted microscope with Continuous Autofocus | Olympus | — | This paper uses a STED Infinity Line system built around an Olympus IX83 inverted microscope, described in Section 4 |
| Lightbox software (V. 16.3.16118) | Abberior | — | Vendor software used for STED image acquisition, described in Section 4 |
| Live Orange Mito dye | Abberior | LVORANGE-0146-30NMOL | Live cell imaging IMM-targeting dye described in Discussion |
| Neurobasal media | Gibco | 21103049 | Used for primary cell materials referred to in Section 2.1.2 |
| Nunc Lab-Tek II 2-well chambered coverglass | Nunc | 155379 | Can purchase a variety of chambers but make sure the coverglass is #1.5 |
| Pasteur Pipets (Fisherbrand) | Thermo Fisher Scientific | 22183632 | |
| Penicillin-Streptomycin (10,000 U/mL) | Gibco | 15140122 | |
| PKmito Orange dye | Spirochrome | SC053 | |
| Poly-D-lysine | Gibco | A3890401 | |
| SH-SY5Y Cell line | ATCC | CRL2266 | |
| Sodium pyruvate (100 mM) | Gibco | 11360070 | Used for primary cell materials described in Section 2 |
| Spectrometer (GENESYS 180 UV-Vis) | Thermo Fisher Scientific | 840309000 | |
| STED Expert Line microscope | Abberior | — | STED setup can be customized, but at time of purchase instrument was considered Abberior’s Expert Line; brief description of setup is available in Section 4 of protocol |
| T25 flask (TC-treated, filter cap) | Thermo Fisher Scientific | 156367 | Other culture vessels and sizes available |
References
- Iovine, J. C., Claypool, S. M., Alder, N. N. Mitochondrial compartmentalization: emerging themes in structure and function. Trends in Biochemical Sciences. 46 (11), 902-917 (2021).
- Gupta, A., Becker, T. Mechanisms and pathways of mitochondrial outer membrane protein biogenesis. Biochimica et Biophysica Acta (BBA) – Bioenergetics. 1862 (1), 148323 (2021).
- Gordaliza-Alaguero, I., Cantó, C., Zorzano, A. Metabolic implications of organelle-mitochondria communication. EMBO Reports. 20 (9), e47928 (2019).
- Klecker, T., Westermann, B. Pathways shaping the mitochondrial inner membrane. Open Biology. 11 (12), 210238 (2021).
- Navarro, A., Boveris, A. The mitochondrial energy transduction system and the aging process. American Journal of Physiology-Cell Physiology. 292 (2), C670-C686 (2007).
- Yu, R., Lendahl, U., Nistér, M., Zhao, J. Regulation of mammalian mitochondrial dynamics: opportunities and challenges. Frontiers in Endocrinology. 11, 374 (2020).
- Horn, A., Raavicharla, S., Shah, S., Cox, D., Jaiswal, J. K. Mitochondrial fragmentation enables localized signaling required for cell repair. The Journal of Cell Biology. 219 (5), e201909154 (2020).
- Glancy, B., Kim, Y., Katti, P., Willingham, T. B. The functional impact of mitochondrial structure across subcellular scales. Frontiers in Physiology. 11, 541040 (2020).
- Bahat, A., et al. MTCH2-mediated mitochondrial fusion drives exit from naïve pluripotency in embryonic stem cells. Nature Communications. 9 (1), 5132 (2018).
- Detmer, S. A., Chan, D. C. Functions and dysfunctions of mitochondrial dynamics. Nature Reviews Molecular Cell Biology. 8, 870-879 (2007).
- Bertholet, A. M., et al. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiology of Disease. 90, 3-19 (2016).
- Zemirli, N., Morel, E., Molino, D. Mitochondrial dynamics in basal and stressful conditions. International Journal of Molecular Sciences. 19 (2), 564 (2018).
- Harwig, M. C., et al. Methods for imaging mammalian mitochondrial morphology: a prospective on mitograph. Analytical Biochemistry. 552, 81-99 (2018).
- Pánek, T., Eliáš, M., Vancová, M., Lukeš, J., Hashimi, H. Returning to the fold for lessons in mitochondrial crista diversity and evolution. Current Biology. 30 (10), R575-R588 (2020).
- Gottschalk, B., Madreiter-Sokolowski, C. T., Graier, W. F. Cristae junction as a fundamental switchboard for mitochondrial ion signaling and bioenergetics. Cell Calcium. 101, 102517 (2022).
- Khosravi, S., Harner, M. E. The MICOS complex, a structural element of mitochondria with versatile functions. Biological Chemistry. 401 (6-7), 765-778 (2020).
- Frezza, C., et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 126 (1), 177-189 (2006).
- Meeusen, S., et al. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell. 127 (2), 383-395 (2006).
- Patten, D. A., et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. The EMBO Journal. 33 (22), 2676-2691 (2014).
- Paumard, P., et al. The ATP synthase is involved in generating mitochondrial cristae morphology. The EMBO Journal. 21 (3), 221-230 (2002).
- Strauss, M., Hofhaus, G., Schröder, R. R., Kühlbrandt, W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. The EMBO Journal. 27 (7), 1154-1160 (2008).
- Basu Ball, W., Neff, J. K., Gohil, V. M. The role of nonbilayer phospholipids in mitochondrial structure and function. FEBS Letters. 592 (8), 1273-1290 (2018).
- Hackenbrock, C. R. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. The Journal of Cell Biology. 30 (2), 269-297 (1966).
- Dlasková, A., et al. Mitochondrial cristae narrowing upon higher 2-oxoglutarate load. Biochimica et Biophysica Acta (BBA) – Bioenergetics. 1860 (8), 659-678 (2019).
- Pérez-Hernández, C. A., et al. Mitochondrial ultrastructure and activity are differentially regulated by glycolysis-, krebs cycle-, and microbiota-derived metabolites in monocytes. Biology. 11 (8), 1132 (2022).
- Mannella, C. A. Structural diversity of mitochondria: functional implications. Annals of the New York Academy of Sciences. 1147, 171-179 (2008).
- Plecitá-Hlavatá, L., Ježek, P. Integration of superoxide formation and cristae morphology for mitochondrial redox signaling. The International Journal of Biochemistry & Cell Biology. 80, 31-50 (2016).
- Scorrano, L., et al. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Developmental Cell. 2 (1), 55-67 (2002).
- Heath-Engel, H. M., Shore, G. C. Mitochondrial membrane dynamics, cristae remodelling and apoptosis. Biochimica et Biophysica Acta (BBA) – Molecular Cell Research. 1763 (5-6), 549-560 (2006).
- Brandt, T., et al. Changes of mitochondrial ultrastructure and function during ageing in mice and Drosophilia. eLife. 6, e24662 (2017).
- Kondadi, A. K., et al. Cristae undergo continuous cycles of membrane remodelling in a MICOS-dependent manner. EMBO Reports. 21, e49776 (2020).
- Quintana-Cabrera, R., Mehrotra, A., Rigoni, G., Soriano, M. E. Who and how in the regulation of mitochondrial cristae shape and function. Biochemical and Biophysical Research Communications. 500 (1), 94-101 (2018).
- Nielsen, J., et al. Plasticity in mitochondrial cristae density allows metabolic capacity modulation in human skeletal muscle: Enlarged mitochondrial cristae density in athletes. The Journal of Physiology. 595 (9), 2839-2847 (2017).
- Afzal, N., Lederer, W. J., Jafri, M. S., Mannella, C. A. Effect of crista morphology on mitochondrial ATP output: A computational study. Current Research in Physiology. 4, 163-176 (2021).
- Heine, K. B., Parry, H. A., Hood, W. R. How does density of the inner mitochondrial membrane influence mitochondrial performance. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 324 (2), R242-R248 (2023).
- Wang, W., Zhao, F., Ma, X., Perry, G., Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Molecular Neurodegeneration. 15, 30 (2020).
- Singh, A., Kukreti, R., Saso, L., Kukreti, S. Oxidative stress: a key modulator in neurodegenerative diseases. Molecules. 24, 1583 (2019).
- Pchitskaya, E., Popugaeva, E., Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium. 70, 87-94 (2018).
- Estes, R. E., Lin, B., Khera, A., Davis, M. Y. Lipid metabolism influence on neurodegenerative disease progression: is the vehicle as important as the cargo. Frontiers in Molecular Neuroscience. 14, 788695 (2021).
- Calkins, M. J., Manczak, M., Mao, P., Shirendeb, U., Reddy, P. H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Human Molecular Genetics. 20 (23), 4515-4529 (2011).
- Petersen, C. A. H., et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proceedings of the National Academy of Sciences of the United States of America. 105 (35), 13145-13150 (2008).
- Gan, X., et al. Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease. 1842 (2), 220-231 (2014).
- Baloyannis, S. J., Costa, V., Michmizos, D. Mitochondrial alterations Alzheimer’s disease. American Journal of Alzheimer’s Disease & Other Dementias. 19 (2), 89-93 (2004).
- Tillement, L., Lecanu, L., Papadopoulos, V. Alzheimer’s disease: Effects of β-amyloid on mitochondria. Mitochondrion. 11 (1), 13-21 (2011).
- Choi, S. Y., et al. C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nature Neuroscience. 22 (6), 851-862 (2019).
- Smith, E. F., Shaw, P. J., De Vos, K. J. The role of mitochondria in amyotrophic lateral sclerosis. Neuroscience Letters. 710, 132933 (2019).
- Costa, V., et al. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Molecular Medicine. 2 (12), 490-503 (2010).
- Costa, V., Scorrano, L. Shaping the role of mitochondria in the pathogenesis of Huntington’s disease: Mitochondrial and Huntington’s disease. The EMBO Journal. 31 (8), 1853-1864 (2012).
- Vanisova, M., et al. Mitochondrial organization and structure are compromised in fibroblasts from patients with Huntington’s disease. Ultrastructural Pathology. 46 (5), 462-475 (2022).
- de Barcelos, I. P., Troxell, R. M., Graves, J. S. Mitochondrial dysfunction and multiple sclerosis. Biology. 8 (2), 37 (2019).
- Park, J., et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 441, 1157-1161 (2006).
- Meng, H., et al. Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nature Communications. 8, 15500 (2017).
- Lu, L., et al. CHCHD2 maintains mitochondrial contact site and cristae organizing system stability and protects against mitochondrial dysfunction in an experimental model of Parkinson’s disease. Chinese Medical Journal. 135 (13), 1588-1596 (2022).
- Cogliati, S., et al. Mitochondrial Cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 155 (1), 160-171 (2013).
- He, B., et al. Mitochondrial cristae architecture protects against mtDNA release and inflammation. Cell Reports. 41 (10), 111774 (2022).
- Polo, C. C., et al. Three-dimensional imaging of mitochondrial cristae complexity using cryo-soft X-ray tomography. Scientific Reports. 10, 21045 (2020).
- Rybka, V., et al. Transmission electron microscopy study of mitochondria in aging brain synapses. Antioxidants. 8 (6), 171 (2019).
- Mannella, C. A. Structure and dynamics of the mitochondrial inner membrane cristae. Biochimica et Biophysica Acta (BBA) – Molecular Cell Research. 1763 (5-6), 542-548 (2006).
- Fry, M. Y., et al. In situ architecture of Opa1-dependent mitochondrial cristae remodeling. biorxiv. , (2023).
- Barad, B. A., Medina, M., Fuentes, D., Wiseman, R. L., Grotjahn, D. A. Quantifying organellar ultrastructure in cryo-electron tomography using a surface morphometrics pipeline. The Journal of Cell Biology. 222 (4), 202204093 (2023).
- Kunz, T. C., Götz, R., Gao, S., Sauer, M., Kozjak-Pavlovic, V. Using expansion microscopy to visualize and characterize the morphology of mitochondrial cristae. Frontiers in Cell and Developmental Biology. 8, 617 (2020).
- Yang, Z., et al. Cyclooctatetraene-conjugated cyanine mitochondrial probes minimize phototoxicity in fluorescence and nanoscopic imaging. Chemical Science. 11 (32), 8506-8516 (2020).
- Liu, T., et al. Multi-color live-cell STED nanoscopy of mitochondria with a gentle inner membrane stain. Proceedings of the National Academy of Sciences. 119 (52), e2215799119 (2022).
- Yang, X., et al. Mitochondrial dynamics quantitatively revealed by STED nanoscopy with an enhanced squaraine variant probe. Nature Communications. 11, 3699 (2020).
- Zheng, S., et al. Long-term, super-resolution HIDE imaging of the inner mitochondrial membrane in live cells with a cell-permeant lipid probe. biorxiv. , (2022).
- Wang, C., et al. A photostable fluorescent marker for the superresolution live imaging of the dynamic structure of the mitochondrial cristae. Proceedings of the National Academy of Sciences of the United States of America. 116 (32), 15817-15822 (2019).
- Feles, S., et al. Streamlining culture conditions for the neuroblastoma cell line sh-sy5y: a prerequisite for functional studies. Methods and Protocols. 5 (4), 58 (2022).
- Shipley, M. M., Mangold, C. A., Szpara, M. L. Differentiation of the SH-SY5Y human neuroblastoma cell line. Journal of Visualized Experiments. (108), e53193 (2016).
- Kovalevich, J., Langford, D., Amini, S., White, M. K. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Neuronal Cell Culture. 1078, 9-21 (2013).
- Biedler, J. L., Helson, L., Spengler, B. A. Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Research. 33 (11), 2643-2652 (1973).
- Swerdlow, R. H. Mitochondria and mitochondrial cascades in alzheimer’s disease. Journal of Alzheimer’s Disease. 62 (3), 1403-1416 (2018).
- Wang, W., Zhao, F., Ma, X., Perry, G., Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Molecular Neurodegeneration. 15, 30 (2020).
- Reddy, P. H. Mitochondrial dysfunction in aging and alzheimer’s disease: strategies to protect neurons. Antioxidants & Redox Signaling. 9 (10), 1647-1658 (2007).
- Horn, A., Raavicharla, S., Shah, S., Cox, D., Jaiswal, J. K. Mitochondrial fragmentation enables localized signaling required for cell repair. Journal of Cell Biology. 219 (5), e201909154 (2020).
- Korecka, J. A., et al. Phenotypic characterization of retinoic acid differentiated SH-SY5Y cells by transcriptional profiling. PLoS ONE. 8 (5), e63862 (2013).
- Baghel, M. S., Thakur, M. K. Vdac1 downregulation causes mitochondrial disintegration leading to hippocampal neurodegeneration in scopolamine-induced amnesic mice. Molecular Neurobiology. 56 (3), 1707-1718 (2019).
- Jiang, S., et al. Mfn2 ablation causes an oxidative stress response and eventual neuronal death in the hippocampus and cortex. Molecular Neurodegeneration. 13 (1), 5 (2018).
- Mu, Y., Gage, F. H. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Molecular Neurodegeneration. 6, 85 (2011).
- Rao, Y. L., et al. Hippocampus and its involvement in Alzheimer’s disease: a review. 3 Biotech. 12 (2), 55 (2022).
- Weerasinghe-Mudiyanselage, P. D. E., Ang, M. J., Kang, S., Kim, J. -. S., Moon, C. Structural Plasticity of the hippocampus in neurodegenerative diseases. International Journal of Molecular Sciences. 23 (6), 3349 (2022).
- . Poly-D-Lysine Available from: https://www.thermofisher.com/order/catalog/product/A3890401 (2023)
- Dravid, A., Raos, B., Svirskis, D., O’Carroll, S. J. Optimised techniques for high-throughput screening of differentiated SH-SY5Y cells and application for neurite outgrowth assays. Scientific Reports. 11, 23935 (2021).
- Hromadkova, L., et al. Brain-derived neurotrophic factor (BDNF) promotes molecular polarization and differentiation of immature neuroblastoma cells into definitive neurons. Biochimica et Biophysica Acta (BBA) – Molecular Cell Research. 1867 (9), 118737 (2020).
- Riegerová, P., et al. Expression and Localization of AβPP in SH-SY5Y cells depends on differentiation state. Journal of Alzheimer’s Disease. 82 (2), 485-491 (2021).
- Hoffmann, L. F., et al. Neural regeneration research model to be explored: SH-SY5Y human neuroblastoma cells. Neural Regeneration Research. 18 (6), 1265-1266 (2022).
- Abiraman, K., Tzingounis, A. V., Lykotrafitis, G. K. Ca 2 channel localization and regulation in the axon initial segment. The FASEB Journal. 32 (4), 1794-1805 (2018).
- E18 Rat Hippocampus. Transnetyx Tissue Available from: https://tissue.transnetyx.com/E18-Rat-Hippocampus_4 (2023)
- Kmito ORANGE – Probe for live cell imaging of mitochondria. Spirochrome Available from: https://spirochrome.com/product/pkmito_orange/ (2023)
- Nyquist Calculator. Scientific Volume Imaging Available from: https://svi.nl/Nyquist-Calculator (2023)
- Segawa, M., et al. Quantification of cristae architecture reveals time-dependent characteristics of individual mitochondria. Life Science Alliance. 3 (7), e2019000620 (2020).
- Arganda-Carreras, I., et al. Trainable Weka Segmentation: a machine learning tool for microscopy pixel classification. Bioinformatics. 33 (15), 2424-2426 (2017).
- Stephan, T., Roesch, A., Riedel, D., Jakobs, S. Live-cell STED nanoscopy of mitochondrial cristae. Scientific Reports. 9, 12419 (2019).
- Erdmann, R. S., et al. Labeling strategies matter for super-resolution microscopy: A comparison between HaloTags and SNAP-tags. Cell Chemical Biology. 26 (4), 584-592 (2019).
