Oral Delivery of Transformed Yeast Cells into the Mouse Intestinal Immune System
Abstract
Source: Hudson, L. E. et al., Transformation of Probiotic Yeast and Their Recovery from Gastrointestinal Immune Tissues Following Oral Gavage in Mice. J. Vis. Exp. (2016)
This video showcases the oral administration of transformed yeast cells into a mouse model. The transformed yeast cells are orally administered using a gavage technique, reaching the mouse's stomach. Later, the cells migrate to the small intestine, where microfold cells engulf them and facilitate their transfer to Peyer's patches and gain access to the intestinal immune system.
Protocol
All procedures involving animal models have been reviewed by the local institutional animal care committee and the JoVE veterinary review board.
1. UV Mutagenesis to Generate Auxotrophic Yeast Strains
- Generate survival curves to determine needed doses of ultraviolet (UV) irradiation
- Prepare YPD (yeast extract peptone dextrose) media and other reagents listed in Table 1 according to standard procedures and inoculate single colonies into 5-10 ml of YPD media. Incubate cultures on a roller drum at 30 °C O/N to saturation for at least 8 hr.
- Determine the cell concentration of overnight (O/N) cultures using a spectrophotometer by diluting cells 1:10 in water in a plastic cuvette. Dilute cells to a concentration of 107 cells/ml in 20 ml of sterile distilled water.
- Pour diluted cells into a sterile plastic Petri dish and, with the lid removed, place the plate 14 cm below a UV bulb.
- Expose cells to serial 5,000 µJ and 10,000 µJ doses of UV irradiation, extracting 500 µl of cells following each increment such that cells are sampled after exposure to 0 µJ, 5,000 µJ, 10,000 µJ, 15,000 µJ, 20,000 µJ, 25,000 µJ, 30,000 µJ, 40,000 µJ, and 50,000 µJ of UV irradiation. Transfer extracted cell samples to sterile 1.5 ml tubes and serially dilute at 1:10 increments in sterile water.
- Pellet cells in each dilution by centrifugation in a microcentrifuge at 16,000 x g for 1 min. Aspirate supernatant and resuspend in a 100 µl volume of sterile water appropriate for plating yeast cells. Pipette the full volume of resuspended cells onto plates containing YPD solid media and use a sterile spreader to distribute cells evenly across each plate.
- Wrap plate edges in Parafilm to prevent drying of media and cover plates in aluminum foil to prevent photo-reactivation and repair of UV-induced mutations. Incubate plates upside down at 30 °C for 2-4 days to allow for growth of viable yeast colonies (Figure 1).
- Count the number of colonies, optionally with the help of a pen to mark off counted colonies, a handheld electronic counter pen, or a counter stand with magnification. Plot as a percentage of total plated cells at each µJ dose of UV irradiation to generate a survival curve for irradiated yeast (Figure 2).
NOTE: Haploid yeast strains can be expected to require lower doses of UV irradiation relative to diploid strains to reach the same percent survival. A strain of yeast lacking functional DNA repair enzymes, such as the rad1 S. cerevisiae mutant, can be used as a positive control to indicate the presence of UV irradiation at very low doses. - Determine the dose of UV mutagenesis to be used for screening by referring to the survival curve established in 1.1.7. The x value of the point along the survival curve where y equals 50 is the UV irradiation dose at which 50% of yeast survive. Screening mutants at this low percent survival may result in a higher yield of successfully mutated strains, particularly for diploid yeast. The 50% survival dose for WT S. boulardii, as shown in Figure 2, is approximately 18,000 µJ.
NOTE: Although such high UV doses increase the risk of mutations in genes for cellular pathways other than the auxotrophic marker gene of interest, this drawback must be balanced against the need to induce mutations in both copies of the auxotrophic marker gene. For haploid strains in which only one gene copy must be mutated, screening at a higher percent survival, such as at 90%, decreases the risk of additional mutations and still allows for sufficient generation of auxotrophic mutants.
- UV mutagenesis and screening for auxotrophic yeast strains
- Prepare yeast as described in steps 1.1.1-1.1.3.
- Expose yeast to the dose of UV irradiation corresponding to 50% survival, as determined in 1.1.8. For WT S. boulardii, this dose was determined to be approximately 18,000 µJ (Figure 2).
- Collect 1 ml volumes of UV irradiated yeast and pellet by centrifugation in a microcentrifuge at 16,000 x g for 1 min. Aspirate supernatant and resuspend cells in 100 µl sterile water.
- Selection of mutants
- If using selection such as with ura3– auxotrophic mutants, plate irradiated yeast onto media containing 5-fluoroorotic acid (5-FOA) to select for cells lacking a functional Ura3 enzyme.
NOTE: Any yeast containing functional copies of Ura3 will convert 5-FOA to the toxin 5-fluorouracil, leading to cell death and allowing for easy selection of ura3-colonies that lack a functional Ura3. Analogous selection approaches are possible for the LYS2 and LYS5; TRP1; and MET2 and MET15 markers using media containing α-aminoadipic acid; 5-fluoroanthranilic acid; and methyl mercury, respectively. - Pipette the 100 µl of resuspended cells onto minimal media containing 5-FOA and use a sterile spreader to evenly coat the plate. Wrap plates in Parafilm and incubate upside down at 30 °C for 2-4 days to allow for growth of viable yeast colonies.
- Confirm the ura3– phenotype of any colony appearing on 5-FOA plates by restreaking onto YPD, uracil– and 5-FOA plates (Figure 3). Use the tip of a sterile toothpick to collect part of a single colony and gently drag the cells across fresh YPD, uracil–, and 5-FOA plates. Again, incubate wrapped plates upside down at 30 °C for 2-4 days.
NOTE: Viable colonies will appear as raised, roughly circular growths while non-viable cells will appear only as an opaque smear without any raised growths (Figure 3).
- If using selection such as with ura3– auxotrophic mutants, plate irradiated yeast onto media containing 5-fluoroorotic acid (5-FOA) to select for cells lacking a functional Ura3 enzyme.
- Screening of mutants
- If generating an auxotrophic mutant for which selection methods are not available, prepare serial 1:10 dilutions of UV irradiated yeast cells in sterile water and pipette the dilutions onto YPD media, using a sterile spreader to evenly coat the plate.
- Wrap plates in Parafilm and incubate upside down at 30 °C for 2-4 days. Determine which dilution allowed for growth of individual colonies that can easily be distinguished from each other, usually no more than approximately 100 colonies per plate.
- Repeat UV mutagenesis of yeast samples as described in 1.2.1-1.2.3 and plate cells at this determined dilution. Pipette the diluted yeast onto YPD media, use a sterile spreader to distribute the cells, and incubate wrapped plates upside down at 30 °C for 2-4 days.
- Screen for auxotrophs by replica plating onto selective media lacking the metabolite of interest. First, secure a sterile velvet pad onto a plate stand and invert the plate with UV irradiated colonies onto the velvet, marking the plate orientation.
- Next, invert a fresh plate lacking the metabolite of interest onto the velvet and lightly press down to transfer cells from the velvet to the plate. Store the original plate at 4 °C. Wrap and incubate the new plate upside down at 30 °C for 2-4 days.
- As an alternative to replica plating, screen mutants by re-streaking colonies from YPD onto selective media. Use the tip of a sterile toothpick to collect part of a single colony and gently drag the cells across a fresh YPD plate and a plate lacking the metabolite of interest. Again incubate wrapped plates upside down at 30 °C for 2-4 days.
NOTE: Care must be taken to select single colonies and streak out colonies multiple times to confirm a homogeneous population of true auxotrophic cells.
- Selection of mutants
- Further confirm the phenotype of irradiated cells by inoculating single colonies into 5-10 ml of both YPD and media lacking the appropriate metabolite (e.g., in uracil– media for a ura3– mutant). Incubate on a roller drum at 30 °C O/N to confirm growth of cells in YPD media but not in the absence of the metabolite.
NOTE: Although growth patterns on solid media should clearly indicate yeast auxotrophic status, it is possible for some yeast to tolerate stresses and form small, slow growing colonies on solid media but yet not tolerate the same conditions in liquid media (unpublished observations). Inoculation into liquid cultures should thus be performed to thoroughly confirm growth patterns of UV irradiated mutants. - For long term storage of confirmed auxotrophic mutants, prepare glycerol stocks by inoculating cells into 10 ml YPD and incubating on a roller drum O/N at 30 °C. Pellet cells by centrifugation for 3 min at 2,500 x g and aspirate media. Resuspend cells in 50% sterile filtered glycerol, transfer to a cryovial, and store at -80 °C.
NOTE: UV mutagenized yeast potentially contain mutations in multiple genes other than in the auxotrophic marker of interest. Before continuing with use of verified auxotrophic mutants, these strains should be further analyzed through gene sequencing and assessment of resistance to pH, bile acid stresses, and other characteristics relevant to probiotic strains, as described elsewhere2. Additionally, use of pcr homology or CRISPR/Cas9 targeting to more selectively mutate auxotrophic markers should be considered as an alternative to UV mutagenesis.
2. Yeast Transformation
- LiOAc Transformation of Yeast
- Inoculate single yeast colonies into 5-10 ml of YPD media and incubate on a roller drum at 30°C O/N.
- To induce log phase growth and increase efficiency of plasmid uptake, determine cell concentration using a spectrophotometer to measure a 1:10 dilution of cells in sterile water in a plastic cuvette. Dilute O/N cultures to an optical density at 600 nm (OD600) of 0.16-0.2 (approximately 2 x 106– 2.5 x 106 cells/ml) in 50 ml of fresh warm YPD and incubate cells on an orbital platform shaker set to 200 rpm until the culture reaches approximately 1 x 107 cells/ml, usually around 4 hr.
NOTE: Transformation efficiency can be measured as a function of the number of successfully transformed yeast colony forming units (CFU) per µg of plasmid DNA. Increased efficiency results in more transformed colonies per µg of plasmid DNA. Subculturing yeast cells and collection during log phase growth is one factor that increases transformation efficiency. - Pellet cells by centrifugation at 2,500 x g for 3 min.
- Aspirate the supernatant and transfer cells to a 1.5 ml microcentrifuge tube by resuspending the pellet in 1 ml sterile water.
- Pellet cells by centrifugation at 16,000 x g for 1 min in a microcentrifuge, aspirate the supernatant and wash cells by resuspension in 1 ml TE/LiOAc (Tris EDTA/lithium acetate) buffer.
- Repeat centrifugation and resuspend cells in TE/LiOAc buffer to a concentration of 2 x 109 cells/ml.
- Prepare transformation mixtures with each of the following: 50 µl of prepared yeast in TE/LiOAc buffer, 5 µl of carrier DNA (10 µg/µl), and 1 µl of plasmid DNA (1 µg). Micrograms of plasmid DNA may be titrated as increasing amounts of DNA may or may not lead to increased transformation efficiency.
NOTE: For a mutant yeast strain lacking one auxotrophic marker, only one plasmid encoding the marker can be transformed per sample. Furthermore, use of a plasmid encoding an easily detectable protein, such as green fluorescent protein (GFP), will allow for efficient determination of proper folding and expression of heterologous protein by the yeast strain subsequent to transformation. - To each preparation, add 300 µl of PEG/LiOAc/TE and vortex thoroughly. Incubation of intact cells with polyethylene glycol (PEG) is essential for efficient transformation.
- Incubate preparations at 30 °C for 30 min with agitation by placing microcentrifuge tubes in a beaker placed onto an orbital platform shaker at 200 rpm.
- Add 35 µl of dimethyl sulfoxide (DMSO) to each reaction and heat shock cells for 15 min in a 42 °C water bath. Although there are conflicting reports of the added benefit of DMSO, heat shock of intact yeast cells has been shown to greatly increase transformation efficiency.
- Wash cells by pelleting via centrifugation as in 2.1.5, aspirating or pipetting off the supernatant, and resuspending in 1 ml sterile water. Gently pipette up and down to break up the cell pellet.
NOTE: It is critical to thoroughly remove the supernatant because the media used in generating competent cells can inhibit yeast growth and colony formation. - Repeat cell pelleting as in 2.1.5, aspirate the supernatant, and resuspend the cells in 100 µl sterile water. Pipette the full volume onto a selective plate and use a sterile spreader to evenly coat the plate with transformed yeast.
- Wrap edges of coated plates in Parafilm to prevent drying of media and incubate upside down at 30 °C for 2 days to allow for growth of transformed yeast cells. Successful, efficient transformation and auxotrophic selection of Saccharomyces cerevisiae yields a high number of colonies per transformation preparation, although yield can be much lower for other strains (Figure 4).
- Store yeast plates for the short term (generally 1-3 weeks) upside down and covered at 4 °C. Prepare glycerol stocks of transformed yeast as described in 1.2.5 for long term storage.
NOTE: Further studies testing transformed CFU are necessary to determine plasmid stability and evaluate proper expression of heterologous protein. Thorough descriptions of plasmid stability, use of immunoblotting to detect denatured proteins recovered from cell samples, enzyme linked immunosorbent assay (ELISA) to detect properly folded three dimensional proteins, and the use of GFP in yeast studies are available elsewhere. Figure 5 shows a representative image of successful GFP expression in transformed S. cerevisiae relative to untransformed cells. Use of such a fluorescent protein is one means of efficiently determining successful heterologous protein production.
- Electroporation of Yeast
- Inoculate single yeast colonies into 5-10 ml of YPD media and incubate on a roller drum at 30 °C O/N.
- Determine cell concentration using a spectrophotometer to measure a 1:10 dilution of cells in sterile water. Dilute O/N cultures in 100 ml fresh warm YPD media to an OD600 equivalent of approximately 0.3. Incubate subcultures at 30 °C on an orbital platform shaker set to 200 rpm until reaching an OD600 of approximately 1.6, usually 4-5 hr. Each 100 ml subculture will generate enough conditioned cells for two transformation reactions.
- Pellet cells by centrifugation at 2,500 x g for 3 min. Aspirate the supernatant and wash cells by resuspending in 50 ml ice cold sterile water. Repeat the wash by pelleting cells, aspirating supernatant, and resuspending in fresh 50 ml ice cold sterile water.
- Pellet the cells again and resuspend in 50 ml ice cold electroporation buffer (1 M Sorbitol, 1 mM calcium chloride (CaCl2)).
- Repeat spin as in 2.2.3, aspirate supernatant, and resuspend cells in 20 ml 0.1 M LiOAc/10 mM dithiothreitol (DTT). Incubate cell suspension on a roller drum at 30 °C for 30 min. Preincubation of cells in LiOAc and DTT synergistically increases the efficiency of electroporation.
- Pellet the cells as in 2.2.3, remove supernatant, and wash by resuspending in 50 ml ice cold electroporation buffer. Repeat centrifugation and resuspend cells in ice cold electroporation buffer to a final volume of 1 ml.
- Prepare on ice: conditioned yeast cells, sterile electroporation cuvettes, and plasmid DNA. Immediately after final resuspension of conditioned cells in 1 ml electroporation buffer, combine 400 µl conditioned yeast cells with approximately 1 µg of plasmid DNA and add to an ice cold 0.2 µm electroporation cuvette. Use of increased amounts of DNA may slightly increase transformation efficiency. Incubate reaction on ice for 5 min, then electroporate with electroporator set to 2.5 kV and 25 µF.
NOTE: As described in 2.1.7, a mutant yeast strain lacking one auxotrophic marker can be transformed with only one plasmid encoding the mutated marker per sample. Also, using a plasmid that encodes an easily detectable protein such as GFP allows for efficient determination of proper folding and expression of heterologous protein subsequent to transformation. - Transfer electroporated cells into 8 ml of a 1:1 mixture of YPD:1 M sorbitol and allow cells to incubate on a roller drum at 30 °C for 60 min.
- Pellet cells as in 2.2.3 and resuspend in 100 µl 1:1 YPD:1 M sorbitol. Plate the full volume onto selective media containing 1 M sorbitol, wrap edges of plates in Parafilm, and incubate plates upside down at 30 °C for 2-5 days to allow for growth of transformed yeast cells.
NOTE: It is critical to test transformed yeast to evaluate proper expression of heterologous protein, as described in 2.1.14 and 2.2.7.
- Oral Gavage of Mice with Transformed Yeast
- Perform all animal care and handling procedures according to the Guide for the Care and Use of Laboratory Animals and Institutional Animal Care and Use Committee approval.
- Prepare O/N yeast cultures by inoculating single colonies of transformed auxotrophic yeast into 5-10 ml of selective media. Incubate cultures O/N for at least 8 hr on a roller drum at 30 °C until saturated.
NOTE: Use of plasmid encoding test proteins such as GFP will allow for ease of protein expression testing in gavaged yeast, as described in 4.7. - For maximal induction of protein expression and to induce log phase growth, prepare subcultures from the O/N cultures by diluting to an OD600 equivalent of approximately 0.16-0.2 in 50 ml of appropriate media as described in 2.1.2.
- Determine the concentration of subcultured cells as in 1.1.2 and adjust to 109 cells/ml. Prepare a 100 µl dose for each mouse, with a few hundred µl extra volume per group to improve accuracy and ease of sample loading.
- Pellet cells by centrifugation at 2,500 x g for 3 min or in a microcentrifuge at 16,000 x g for 1 min. Aspirate supernatant and resuspend cells by adding an equal volume of sterile water and gently pipetting up and down.
- Fix an appropriate gauge gavage needle (22 G for 15-20 g mice) onto a 1 ml sterile syringe and load yeast sample, being sure to eliminate any bubbles and set plunger to a 100 µl increment. Load an additional syringe with sterile water to gavage control mice and check for presence of any contaminating yeast.
- Pick up the mouse to be gavaged using the non-dominant hand, with index finger and thumb tightly grasping the skin around the neck (Figure 5A). Tuck the tail under the small finger to prevent movement of the lower body. Be sure that the grip is secure and prohibits the mouse from moving its head in order to prevent damage to internal tissues during gavage.
NOTE: Estimate how far the gavage needle should be inserted by holding the needle against the mouse such that the bulb is even with the xiphoid process of the sternum. Inserting this length of the needle will typically allow the gavage needle bulb to enter the stomach. - Using the dominant hand, gently insert the gavage needle into the mouse esophagus by angling the needle along the roof of the mouth and back of the throat, keeping slightly to the left of center. Wait for the mouse to swallow the bulb of the needle and allow the needle to descend slightly further to the point estimated in 3.7 (Figure 5B). If any resistance is felt during insertion of the gavage needle or if the mouse at any time begins to gasp, gently remove the needle and again try to find the esophagus.
- After the mouse has swallowed the bulb of the gavage needle, gently depress the syringe plunger to administer 100 µl (108 CFU) of yeast directly into the mouse stomach.
NOTE: Although mice are unable to vomit any of the gavaged solution after administration, it is possible for reflux to occur during gavage. Proper insertion of the gavage needle, as well as adjusting the volume and viscosity of the solution, can help to limit reflux and ensure accurate dosing. - Carefully remove the gavage needle from the mouse stomach and esophagus and return the mouse to the cage. Check that the mouse is breathing and moving normally after gavage to ensure that the gavage needle was properly inserted throughout the procedure and that no solution was aspirated.
Table 1. Reagents List. Described are the reagents needed for making each of the solutions, yeast media and plates, and transformation buffers used for the protocols in this manuscript.
| Solutions | Yeast Media and Plates | Transformation Reagents |
| Polyethylene glycol (PEG) 50%: | YPD: | TE/LiOAc: |
| 250 g PEG 3350 | 20 g peptone | 50 ml 10x TE |
| 500 ml sterile water | 20 g dextrose | 50 ml 10x (1M) LiOAc |
| Filter sterilized | 10 g yeast extract | 400 ml sterile water |
| 1 L water | Filter sterilized | |
| Autoclave | ||
| TE 10x: | YPD plates: | PEG/TE/LiOAc: |
| 100 mM Tris | 20 g peptone | 400 ml 50% PEG |
| 10 mM EDTA | 20 g dextrose | 50 ml 10x TE |
| pH to 7.5 and filter sterilize | 20 g agar | 50 ml 10x (1M) LiOAc |
| 10 g yeast extract | ||
| 1 L water | ||
| Autoclave | ||
| 20% glucose: | Uracil– selective media | Carrier DNA (SS DNA): |
| 200 g dextrose | 2 g amino acid mix lacking uracil | Store at -20 °C and prior to use heat for 1-2 min at 100 °C to melt strands and store on ice |
| 1 L water | 6.7 g yeast nitrogen base without amino acids | |
| Filter sterilized | 1 L water | |
| Sterilize by autoclaving or sterile filtering | ||
| Add 20% glucose 1:10 before use | ||
| 50% glycerol: | Uracil– plates: | Electroporation buffer: |
| 500 ml glycerol | In a 250 ml flask: | 1 M Sorbitol |
| 500 ml water | 2 g amino acid mix lacking uracil | 1 mM CaCl2 |
| Autoclave | 6.7 g yeast nitrogen base without amino acids | Fill with distilled water |
| 150 ml water | Autoclave and store at 4 °C | |
| In a 2 L flask: | ||
| 20 g agar | ||
| 750 ml water | ||
| Autoclave flasks separately, then mix together with 100 ml 20% glucose | ||
| Complete IMDM | 5-FOA+ plates: | LiOAc/DTT |
| 500 ml Iscove's Modified Dulbecco's Media | Autoclave in a 2 L flask: | 0.1 M LiOAc |
| 5 ml penicillin streptomycin glutamine 100x | 20 g agar | 10 mM DTT |
| 500 μl 2-mercaptoethanol | 750 ml water | |
| 10% heat inactivated fetal bovine serum | Mix: | |
| 2.5 ml sodium pyruvate 100 mM | 6.7 g yeast nitrogen base without amino acids | |
| 2 g amino acid mix without uracil | ||
| 150 ml warm water | ||
| When cool, add: | ||
| 0.05 g uracil powder | ||
| 1 g 5-FOA | ||
| Stir and filter sterilize | ||
| Add to autoclaved agar solution | ||
| Mix with 100 ml 20% glucose |
Representative Results

Figure 1. Yeast colonies grown on YPD media. Example YPD plate showing viable colony forming units (CFU) of probiotic yeast after UV irradiation. Cells were serially diluted such that individual CFU can be distinguished and counted.
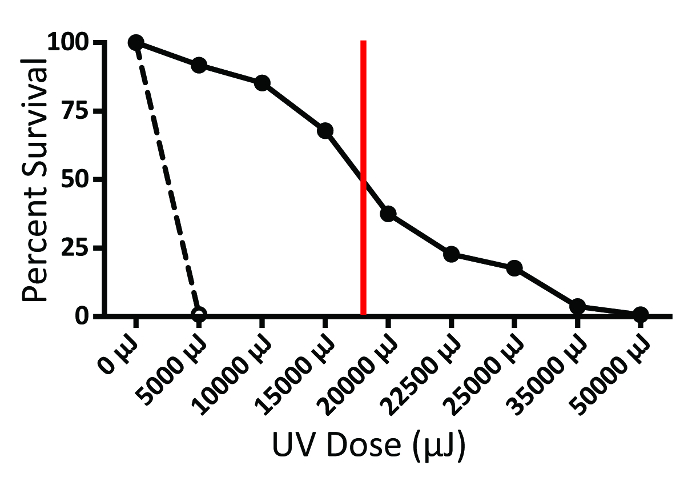
Figure 2. Survival curve for diploid probiotic yeast. Number of viable S. boulardii CFU as a percent of total plated cells was plotted for each µJ dose of UV irradiation (solid line). The vertical red line indicates the µJ UV dose corresponding to 50% survival of this yeast strain. A rad1 S. cerevisiae mutant, which cannot repair damage from UV mutagenesis, is shown as a control (dashed line).
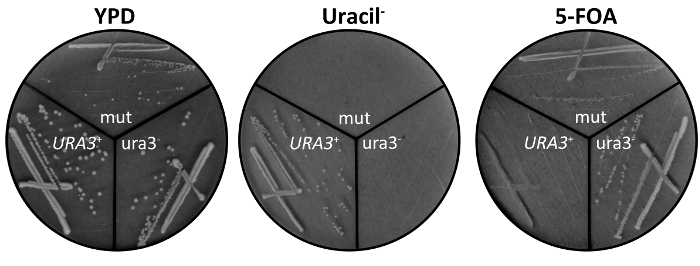
Figure 3. Confirmation of ura3– phenotype of UV irradiated cells on YPD, uracil–, and 5-FOA plates. Cells from individual UV mutant colonies were collected using the tip of a sterile toothpick and gently streaked across YPD, uracil–, and 5-FOA plates. Cells were first streaked in two perpendicular crossing lines, then a new toothpick was used to pass through the second line and continue spreading cells until individual cells separate. A true ura3– mutant (mut) grows on YPD media and in the presence of 5-FOA, but not in the absence of uracil. Control ura3– S. cerevisiae (ura3–) and URA3+ S. boulardii (URA3+) are shown for comparison and to confirm proper preparation of yeast media.
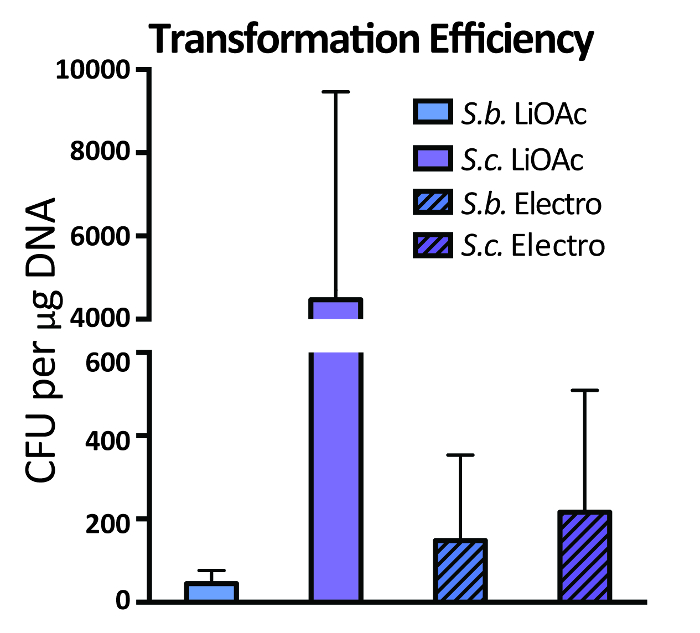
Figure 4. Transformation Efficiency of Saccharomyces strains. Wild type S. boulardii (S.b.) and a laboratory S. cerevisiae strain (S.c.) were transformed using the described LiOAc (LiOAc) and electroporation (Electro) protocols. Results are plotted as mean CFU obtained per µg of plasmid encoding a kanamycin resistance marker. Bars show the mean of duplicate experiments with error bars depicting the standard error of the mean.
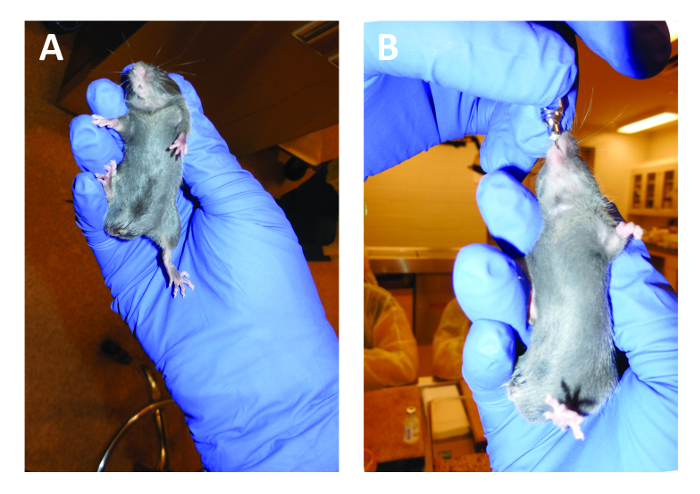
Figure 5. Proper handling of a C57BL/6 mouse for oral gavage. The mouse is held tightly in the non-dominant hand with the tail tucked under the small finger so that no movement is possible (A). The gavage needle is inserted into the pharynx along the roof of the mouth. The mouse is allowed to swallow the bulb of the gavage needle, allowing the solution to then enter the stomach as the plunger is depressed (B).
Offenlegungen
The authors have nothing to disclose.
Materials
| SmartSpec 3000 Spectrophotometer | BioRad | 170-2501 | Example of spectrophotometer for determining cell concentration and OD600 of yeast cultures |
| New Brunswick Roller Drum | Eppendorf | M1053-4004 | Example of roller drum for yeast culture incubation |
| UV Stratalinker 2400 | Stratagene | 400075-03 | Example stratalinker |
| Stuart Colony Counter SC6PLUS | 11983044 | Fisher Scientific | Plate stand with magnification records colony count upon sensing pressure from pen |
| Scienceware Colony Counter | F378620002 | Bel-Art Scienceware | Hand held colony counter pen |
| Replica plating device | Fisherbrand | 09-718-1 | Example of replica plating stand and pads |
| Velveteen squares | Fisherbrand | 09-718-2 | |
| L shaped sterile cell spreaders | Fisherbrand | 14665230 | |
| Deoxyribonucleic acid, single stranded from salmon testes | Sigma-Aldrich | D7656-1ML | Example carrier DNA for yeast LiOAc transformation |
| Gavage needles | Braintree Scientific | N-PK 002 | For mice 15-20 g, the suggested needle is a 22 gauge (1.25 mm ball), 1 in long, straight reusable gavage needle. For mice weighing greater than 20 g, 20 gauge or larger straight or curved gavage needles may be used |
| 1mL sterile slip-tip disposable tuberculin syringe | Becton Dickinson | BD 309659 | |
| Blunt forceps such as Electron Microscopy Sciences 7" (178 mm) serrated tip, broad grip forceps | Electron Microscopy Sciences | 77937-28 | Example of blunt forceps needed for dissection |
| Straight and curved dissection scissors | Electron Microscopy Sciences | 72966-02 and 72966-03 | Examples of scissors needed for dissection |
Tags

.