1. Preparation of the Crab Stomatogastric Nervous System
Adult Cancer pagurus L. were obtained from local sources (Newcastle University, Dove Marine Laboratories) and kept in filtered, aerated seawater (10 – 12°C). Animals were anesthetized by packing them in ice for 20 – 40 min before dissection. We used isolated stomatogastric nervous systems (STNS)1. The STNS was pinned down in a silicone elastomer-lined (ELASTOSIL RT-601, Wacker, Munich, Germany) Petri dish and continuously superfused (7 – 12 mL/min) with chilled saline (10-13°C). Physiological saline consisted of (mMol*l-1): NaCl, 440; MgCl2, 26; CaCl2, 13; KCl, 11; trisma base, 10; maleic acid, 5. Saline was kept at a constant temperature of 11 – 13°C and at pH 7.4 – 7.6.
The detailed steps of the STNS dissection and preparation, including the desheathing of the stomatoastric ganglion (STG), are presented in the JoVE article by Guttierez and Grashow1. All experiments were carried out in accordance with the European Communities Council Directive of 24th November 1986 (86/609/EEC). Figure 1A shows a schematic diagram of the STNS and Figure 1B shows a typical desheathed STG having its neurons arranged as a flat semicircle in the posterior part of the ganglion.
The Petri dish with the STNS is fixed to the operating platform of the imaging microscope (BW51 WI; Olympus, Tokyo, Japan) with modelling clay. Both, the microscope stage and the microscope are mounted on a vibration isolated table (Scientifica, Uckfield, UK) to prevent movement artefacts during the optical recording. The motor patterns generated in the stomatogastric ganglion (STG) are monitored using extracellular recordings2-4. This is done through the following steps:
- A petroleum jelly-based cylindrical compartment is built around a section of the main motor nerve (lvn) to electrically isolate the nerve from the bath (this is done on the dissection stage before the Petri dish is placed on the operating platform of the imaging microscope).
- One of two stainless steel electrode wires is placed in this compartment, the other one in the bath as a reference electrode.
- The differential signal is recorded, filtered and amplified with an AC differential amplifier (Univ. Kaiserslautern, Germany).
- The motor activity of the ganglion is monitored using an oscilloscope (DL708E; Yokogawa, Tokyo, Japan) and is additionally recorded using a data acquisition board (CED Power, 1401, Cambridge Electronic Design, Cambridge, UK) and the Spike 2 v6.10 software (Cambridge Electronic Design, Cambridge, UK). Files are analyzed using Spike 2 v6.10 (Cambridge Electronic Design, Cambridge, UK).
2. Preparation of the Dye Solution
We used the fluorescent voltage-sensitive dye Di-8-ANEPPQ (Cambridge Bioscience)5, which has a double positive charge that facilitates the loading of the dye through microelectrodes by using positive current pulses. The dye solution should be protected from light as much as possible. The dye solution is prepared as follows:
- 5 mg of dye is mixed with 1 mL of F-127 Pluronic acid in DMSO (Invitrogen) solution.
- The plastic vial containing the dissolved dye is centrifuged for 20 minutes at 12,000 rotation/minute in a Sigma Microcentrifuge 1-14 (Sigma, Osterode, Germany) in order to separate larger dye particles that may clog the microelectrode.
- The supernatant (top 200 μL) of the dye solution is separated using a pipettor and stored in a separate plastic vial. Both vials are wrapped in aluminium foil to prevent exposure to light.
- The dye solution is frozen at -20 °C.
- Before use the dye solution is melted by placing the closed and wrapped vial in warm (25 °C) water for 10-15 minutes.
3. Dye Loading by Intracellular Injection using Sharp Microelectrodes
We used sharp microelectrodes to load the neurons with the dye. To check the dye loading we use the MiCAM02 imaging system (SciMedia, Tokyo, Japan)6. The imaging system has two CCD cameras: the HR camera has a larger sensor chip (6.4 mm x 4.8 mm) providing better spatial resolution with the best temporal resolution being 1.4 ms, the HS camera has a smaller (2.9 mm x 2.1 mm) but faster sensor chip allowing faster imaging having 0.7 ms as its best temporal resolution. The steps of the dye loading are as follows:
- Microelectrodes are pulled with a P-97 Flaming – Brown (Sutter Instruments, Novato, CA, USA) electrode puller using 10 cm long, 1 mm outer diameter glass tubes with filament (GB100TF 8P, Science Products, Hofheim, Germany). The electrodes are pulled to have a resistance in the range of 25 – 40 MOhm in 3M KCl (for appropriate pulling parameter settings follow the pipette cookbook provided by the maker of the electrode puller).
- The frozen dye solution is molten and a very small amount of it is sucked up into a microfil needle (MicroFil – 34G, World Precision Instruments, Sarasota, FL, USA) – the sucked up dye fills around 2/3rd of the microfil needle.
- The dye solution in the microfil needle is injected into the tip of a microelectrode. The microelectrode is placed in a dark box for 15 minutes.
- The microelectrode is backfilled with 3M KCl solution. The electrode is then placed back into a dark electrode storage box for another 10-20 minutes to allow the dye solution to reach the fine tip of the electrode by the suction of the capillary force acting in the microelectrode.
- The ready microelectrode is placed into an electrode holder and fixed onto the headstage of the intracellular amplifier (IA-251A, Warner Instruments Corporation, Hamden, CT, USA).
- The intracellular amplifier is connected to the data acquisition board that provides a pulse signal to drive current pulses into the microelectrode.
- The microelectrode is placed gently into the membrane of an STG neuron using a microelectrode manipulator (PatchStar, Scientifica Ltd, Uckfield, UK). Since the working distance of the 10x objective (UMPLFL10XW, NA 0.30, WD 3.3mm; Olympus Corporation, Tokyo, Japan) of the imaging microscope is very small, the microelectrode should be positioned in an oblique position (around 30°) and the movement of the microelectrode into the right position requires several steps of re-focusing of the microscope objective and moving the microelectrode. To start the microelectrode is moved in a position such that its tip is above the STG. The microscope objective is lowered until the electrode tip appears in focus. Then it is lowered further but not as much to touch the microelectrode. The microelectrode is lowered until it re-appears in the focus of the objective. These steps are repeated until the neurons appear clearly in the focus plane of the objective. Then the microelectrode is lowered until it touches and gently pierces the membrane of the selected STG neuron. This is monitored using the oscilloscope.
- The intracellular electrode is used to record the activity of the selected STG neuron in order to identify the neuron given its activity profile and the relationship between its activity and the STG activity recorded from the extracellular electrode2,3,7.
- The intracellular amplifier is switched into current injection mode and is used for 30 minutes to inject 10 nA positive current steps into the microelectrode that last for 1 second and are separated by 1 second gaps with no current injected into the microelectrode. The injected current drives the positively charged dye molecules into the recorded neuron. If equipment is available multiple neurons may be injected simultaneously.
- Ten minutes into the injection period and after the end of the injection the dye spread within the neuron is checked. To do this we use the MiCAM02 imaging system (SciMedia Ltd, Tokyo, Japan) with the 10x objective and illumination by the ultra-low ripple halogen light source (HL-151; Moritex, Tokyo, Japan) through the filter cube MSWG2 (with 480-550 nm excitation filter; Olympus Corporation, Tokyo, Japan). The imaging system is set to use the HR camera with 192 x 128 pixel resolution, 40 ms imaging time, the ‘hbin’ mode, and 10 frames per imaging session. If the dye filling is successful the imaging should show a dyed patch in the neuron around the location of the microelectrode, and this dyed patch should grow between the images taken after 10 minutes and after 30 minutes. If the spread of the dye is insufficient after 30 min, more injection time is granted. Alternatively, a new dye electrode may be used.
- The electrode is pulled out of the neuron and the neuron is left to relax for at least 20-30 minutes. Within this time, the next neuron may be injected with dye.
4. Imaging of Dyed Neurons
The activity of the single neurons can be imaged after the dye filling procedure. Alternatively two or more neurons may be filled to image the simultaneous activity of several STG neurons. In principle many and possibly all STG neurons can be filled with dye. The imaging of the neurons is done with the MiCAM 02 imaging system using the HR camera. The procedure of the imaging is as follows:
- The microscope objective is changed and a high light transmission efficiency 20x objective (XLUMPLFL20XW/0.95, NA 0.95, WD 2.0mm; Olympus Corporation, Tokyo, Japan) is used for the imaging data collection. This objective collects much more light that the 10x objective and also gives higher magnification providing more detailed image of the neurons. Being more light efficient means that small amount of dye loading is already visible with this objective and also that we can use lower light intensity for the imaging reducing the potential photo damage that may be caused to the neurons by the dye.
- The microscope’s operating stage is positioned such that the neurons that are meant to be imaged are in the field of view of the objective and the objective is focused onto them.
- The extracellular recording data is also fed into the MiCAM 02 imaging system through its analogue input channel. This allows the relatively easy relating of optical signals and corresponding motor patterns or extracellularly recorded spikes in the analysis phase.
- The MiCAM 02 system is set to use 96 x 64 pixel resolution with 2.2 ms imaging time or 48 x 32 pixel resolution with 1.5 ms imaging time. In both cases the ‘hbin’ setting may be used if the dyeing of the neurons is not very strong.
- The light level is set as low as possible (i.e. the dye in the neurons should generate still sufficient fluorescence for the imaging) to avoid photo modulation and photo damage of the recorded neurons.
- The room lighting and all remaining light sources are turned off. In addition, a black curtain may be closed around the recording rig to prevent light exposure from external sources.
- The imaging sessions are set to be between 4 – 16 seconds long.
- The spontaneous activity of the dyed neurons is recorded over several imaging sessions.
5. Analysis of the Optical Imaging Data
To analyse the imaging data we used the BVAna software (version 10.08; SciMedia Ltd, Tokyo, Japan) associated with the MiCAM 02 imaging system. The software supports the visualisation of the imaging data and provides a range of analysis tools as well. The steps of the data analysis and interpretation are as follows:
- An appropriate circular region of interest is selected on each recorded neurons. Typically we use 2 – 3 pixel radius regions of interest for investigating neurons at the 48 x 32 pixel resolution, and 6 – 8 pixel radius regions of interest for investigating neurons at 96 x 64 resolution.
- The temporal smoothing of the data may be necessary, especially if the dye generates relatively low amount of fluorescence (low background value in the data indicates this; it may be due to the low light level that is used or low level dye loading, for example in a neurite of the recorded neuron). Typically in such cases we used temporal smoothing with 3 – 5 time units.
- In addition to the optical recordings we also display the extracellular recording by selecting to display the analogue input of the imaging system.
- The parameters for the visualisation of the optical data and the analogue input should be set appropriately in order to see all recordings on the computer screen.
- Considering the optical recordings of the neurons, the extracellular recording from the lvn, and the classification of the neuron based on the earlier intracellular recording, it is possible to determine the identity of the neuron and to relate its activity to the neural activities recorded from the nerve.
- It is expected that the optical recordings show clearly, and without the need of averaging over multiple similar events, the neural activities corresponding to extracellularly recorded spikes and also subthreshold events such as inhibitory post-synaptic potentials caused by the activity of other neurons.
- The data export features of the BVAna software can be used to export the optical recordings calculated for the selected regions of interest and also the analogue input that contains the recording from the lvn. The exported data can be processed and analyzed further using other data analysis software as well (e.g. in the simplest case Microsoft Excel (Microsoft, Redmond, WA, US) may be used for additional visualisation and data processing).
6. Representative Results
Figure 2 shows the simultaneous recording of two neurons (LP, lateral pyloric neuron; PD, pyloric dilator neuron) in the pyloric central pattern generator of a crab STG together with the recording from the lvn. The optical recordings of the LP and PD neurons identified on the basis of their intracellular recordings show that indeed these match well with the extracellular recordings corresponding to LP and PD neurons. There is a consistent 12 ms delay between the optical and lvn recordings of spike peaks (see inset of Figure 2) due to the axonal transmission delay between the ganglion and the extracellular recording site. The presented data also shows that the neural activity in the soma that corresponds to spikes of the recorded neurons is clearly detectable.
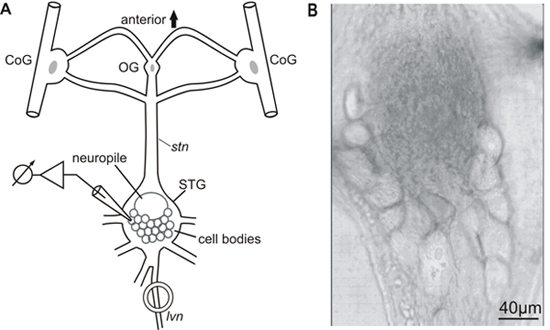
Figure 1. A) Schematic diagram of the stomatogastric nervous system (STNS). CoG, commissural ganglion; OG, esophageal ganglion; STG, stomatogastric ganglion; stn, stomatogastric nerve; lvn, lateral ventricular nerve. B) The crab stomatogastric ganglion (STG) – the image shows the semicircular posterior arrangement of STG neurons.
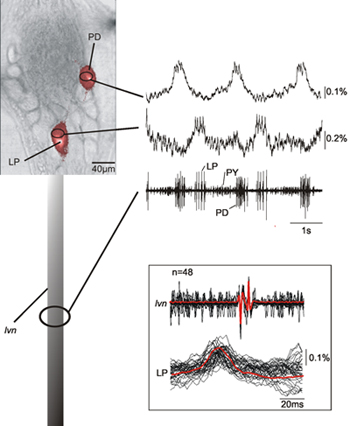
Figure 2. Simultaneous single-sweep recording of a PD and an LP neuron together with the recording of the lvn. The data shows that that the optical and nerve recordings match very well and that we can identify the neural activities corresponding to spikes recorded form the nerve. The inset shows an overlay of several sweeps of LP action potentials – both optically and electrically recorded – and their average demonstrating the match between optically and electrically recorded activities. Due to the fact that our optical recording method does not filter out the low frequency components of the data we are also able to record slow membrane potential changes characteristic for PD and LP neurons14.
