Anterograde Transport
Application of this protocol to infections of dissociated SCG cultures with PRV 348, a recombinant PRV strain expressing GFP-Us9 and gM-mCherry membrane fusion proteins, has facilitated the visualization of the anterograde transport of virions (Figure 3 and Supplemental Movie 4). Incorporation of these fusion proteins into viral particles results in their detection on transporting puncta, and the aforementioned imaging conditions minimize the offset of each fluorophore on a moving particle during filter switching. In a three-minute imaging window, numerous transporting puncta are commonly observed and large sample sizes for colocalization analyses are rapidly generated. We have previously quantified the colocalization of GFP-Us9, the alphaherpes virus axonal targeting protein, with other structural proteins using this approach 11. Similar analyses of other dual recombinant PRV strains have also been described 11, and recent work has documented the co-transport of PRV proteins with a fluorescently tagged host motor protein in a further extension of this protocol 21. Utilizing fast switching filter wheels and paired multi-pass dichroic mirrors allows rapid sequential acquisition of two or more fluorescent channels. We can currently achieve two-channel sequential acquisition at speeds of up to 0.8 frames per second. For single channel video microscopy, which is limited only by the fluorescence exposure time and read-time of the camera, we can achieve acquisition rates greater than 6 frames per second.
Anterograde Spread
To visualize anterograde spread events, areas of interest are visualized every 5 to 10 min for a 20 hr period, during which time virions will transport down and egress from axons, infecting the nearby epithelial cells. A representative frame from the large image area is presented in Figure 4A and Supplemental Movie 5. Note the spatially separated nature of the fluorescent protein expressing cells, indicative of primary infection from axons. Infected cells can be tracked backwards during the movie to visualize the fluorescent capsids that initiated the viral infection. These individual cells can be isolated from the large frame and tracked without a significant loss of detail. The representative frame (Figure 4B) from Supplemental Movie 6 depicts a time after virions have deposited the fluorescent capsid into the cytoplasm.
Data Analysis
A range of different analyses can be performed on live-cell imaging datasets including particle tracking, colocalization studies, and quantification of virion spread events. We often perform our analyses directly in the NIS Elements software or utilizing a plug-in extension available for the ImageJ software package that enables .nd2 file type support. Different hypotheses and experimental setups necessitate unique analytical methods. Our group has described methods for assessing colocalization of fluorescent proteins on dynamic structures 11,21 and time-lapse quantification of individual particle spread events 10,12,22. The methodologies describe in this manuscript and our previous studies are flexible and can be applied to different viral infections as well as the transport of cellular structures, e.g. mitochondria 23.
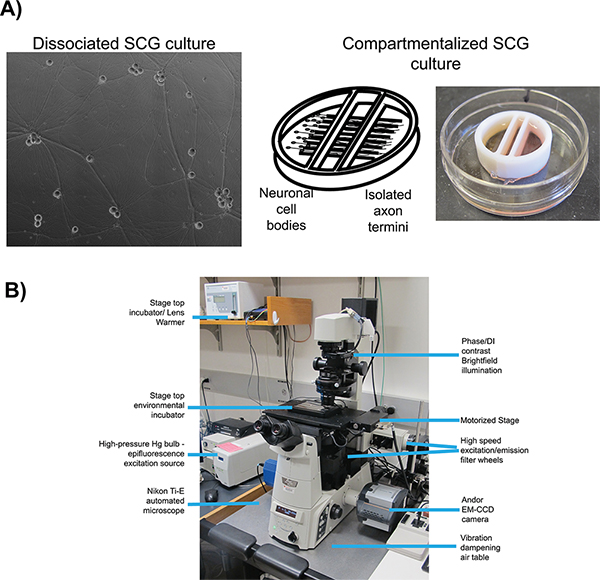
Figure 1. Schematic of microscopy set up for live cell imaging. A) Culture types employed for studies of anterograde transport (dissociated) and anterograde spread (chambered) respectively. Panel 1 is a phase contrast image of a mature, dissociated Superior Cervical Ganglia (SCG). Panel 2 is a schematic of the compartmentalized culture system, pictured in panel 3, with SCG neurons plated in the left compartment with axonal projections extending beneath the internal barriers into the far right compartment. B) The inverted epifluorescent microscope and stage top incubator configuration used for video microscopy experiments. Click here to view larger figure.
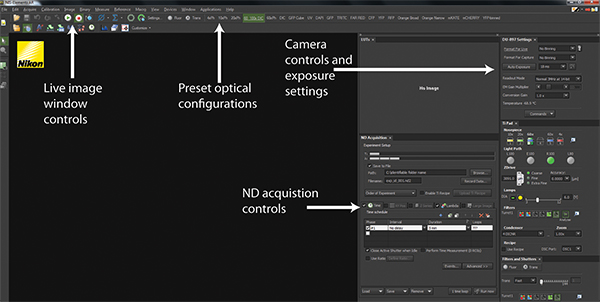
Figure 2. NIS-Elements software interface. A screenshot of a standard user interface in NIS-Elements is displayed. The Play and Stop buttons control the live-image window depicting the current hardware configurations for imaging as well as what is currently being imaged. Across the top panel are a series of Optical Configurations that implement preset hardware settings to configure the microscope for trans-illuminated or fluorescence detection. The Camera control and exposure settings allow configuration of camera detection to optimize image quality and speed. All experiments are coordinated using ND Acquisition controls. Click here to view larger figure.
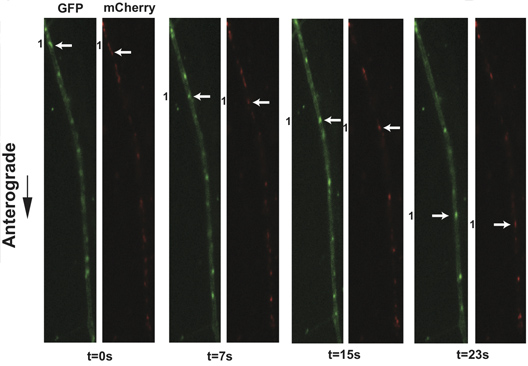
Figure 3. Live cell imaging of SCG axons at 8 hr post-infection with PRV 348. PRV 348 infected neurons express the GFP-Us9 and gM-mCherry membrane fusion proteins. Anterograde-transporting structures within distal axons are visualized in two fluorescent channels, GFP and mCherry. An anterograde-transporting structure (white arrow) progresses within the axon as depicted in the sequential images taken from Supplemental Movie 4. The two fluorescent proteins are spatially off-set due to sequential acquisition of fluorescent channels and the rapid movement of viral structures. Click here to view larger figure.
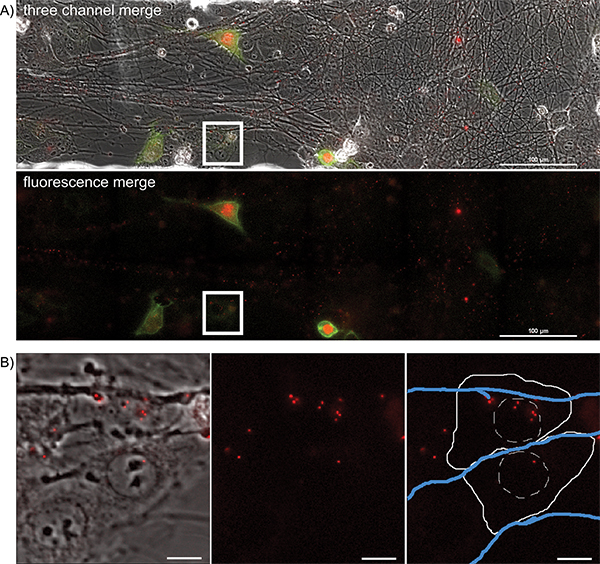
Figure 4. Overnight timelapse imaging of anterograde spread events from infected axons to epithelial cell clusters during a multi-color infection. Representative images from a single position, large image tiled time-lapse movie of virion transmission from PRV 427 infected axons into recipient epithelial cells. A) A merged image of the transmitted, YFP and RFP fluorescent images at a time post infection where recipient cells are beginning to express the YFP-CAAX and VP26-mRFP fusion proteins. In the second panel, the YFP and RFP channels are shown together. Note the large number of mRFP puncta associated with the axon tracts. These are assembled virions undergoing long distance transport within axons (Supplemental Movie 2). The white box represents the area of interest highlighted in part B. B) Representative image of recipient epithelial cells containing infecting capsid assemblies (See also Supplemental Movie 3). Panel 1 is a merge of the transmitted and RFP channels. Panel 2 is the RFP channel alone. Panel 3 is the RFP channel with a schematic representation of the cell outline (solid white line), nucleus (hashed white line), and axons (blue lines). Click here to view larger figure.
Supplemental Movie 1. Software settings for rapid, sequential acquisition of axonal virions. The steps associated with determining exposure settings, configuring ND acquisition controls, and acquiring a rapid frame acquisition movie of fluorescent virions undergoing axonal transport is presented. The SCG neuron culture was infected with PRV 427 eight hours prior to imaging. Click here to view movie.
Supplemental Movie 2. Software settings for overnight imaging of axon-to-cell spread. The steps associated with configuring ND acquisition controls for overnight imaging of axon-to-cell spread events are presented. Click here to view movie.
Supplemental Movie 3. Conversion of .nd2 files into presentable movie formats. The steps associated with converting the raw data .nd2 files into either .avi or .mov file formats for presentation using common video players. Click here to view movie.
Supplemental Movie 4. Live cell imaging of SCG axons at 8 hr post-infection with PRV 348. Click here to view movie.
Supplemental Movie 5. Large image area of overnight imaging of axon-to-cell spread. Click here to view movie.
Supplemental Movie 6. Enlarged area of overnight timelapse imaging of axon-to-cell spread. Click here to view movie.
| Strain | Genotype | Utility |
| PRV 341 | GFP-Us9 (membrane) / mRFP-Vp26 (capsid) | Virion transport experiments11 |
| PRV 348 | GFP-Us9 (membrane) / gM-mCherry (membrane) | Virion transport experiments11 |
| PRV 427 | mRFP-VP26 (capsid)/ YFP-CAAX (cellular plasma membrane) | Anterograde spread experiments10 |
Table 1. Useful recombinant PRV strains expressing fluorescent fusion proteins.
| Name | Company | Catalogue number |
| 35 mm glass bottom culture dish | MatTek Corporation; Ashland, MA | P35G-1.5-20-C |
| 35 mm μ-Dish | Ibidi USA LLC, Verona, WI | 81156 |
| Microscope body | Nikon Instruments Inc. | Nikon Eclipse Ti |
| Motorized microscope X-Y stage | Prior Scientific; Rockland, MA | H117 ProScan Flat Top |
| Motorized filter wheels | Prior Scientific | HF110 10 position filter wheel |
| Fluorescent illumination source | Lumen Dynamics; Mississauga, Ontario Canada | X-Cite 120Q |
| Multi-band Fluorescence filter sets | Chroma Technology Corp.; Bellows Falls, VT | 89000 Sedat Quad – ET 89006 ECFP/EYFP/mCherry – ET |
| EM-CCD camera | Andor Technology USA; South Windsor, CT | iXon3 897 |
| Chamlide stage top environmental incubator | Live Cell Instruments; Seoul, South Korea | TC-L-10 |
| Objective lens heater | Bioptecs; Butler, PA | 150803 Controller 150819-12-08 Heater |
| Analysis software | Nikon Instruments Inc.; Melville, NY | NIS Elements |
Table 2. Specific reagents and equipment.
