To facilitate data interpretation and presentation, care should be taken not to damage the embryos during collection and manipulation, so that all cells and their relative position can be analyzed. Figure 2A – D shows examples of intact blastocysts at different stages with an expanded cavity. Should damage occur, extra care should be taken when analyzing and interpreting results.
The quality and reliability of the data generated using this protocol is dependent on the quality of the fixation and on the signal-to-noise ratio of the antibodies used to detect the proteins of interest. Always use fresh fixative and test new antibodies, with appropriate positive and negative controls, before beginning a set of experiments. Figure 2A shows examples of good antibodies for a number of nuclear proteins. Figure 2B shows an example of a good antibody for a cytoplasmic protein (DAB2). Figure 2C shows an example of a bad staining, with low signal-to-noise ratio, where the sample was fixed for only 10 min. In this case, the antibody used for GATA4 (Santa Cruz, sc-1237) requires O/N fixation to provide a strong signal (see 24 for instances of embryos fixed O/N and stained with this antibody). Increasing the signal level during post-processing reveals a very noisy image. By contrast the anti-GATA4 used for Figure 2A (sc-9053) provides high signal-to-noise ratio after only 10-min fixation. Details for these and other robust antibodies are provided in the Materials section.
The limiting factors for a good MINS segmentation are a) the magnification of the objective used for imaging, b) the Z-resolution and c) the quality of the nuclear staining. Figure 2D shows examples of embryos imaged with an oil immersion 40X objective with a NA of 1.30 and a 0.21 mm WD. Middle panels show magnifications of the ICMs where individual nuclei and the border between them can be distinguished. If not using DNA staining (i.e., Hoechst, DAPI, TO-PRO3, YO-YO1, etc.) fluorescence values for quantification, acquisition parameters can be individually adjusted to obtain the best signal. Bottom panels show how the more advanced the embryo, the higher the nuclear density and thus the higher the chance of segmentation errors (arrowhead and arrow). Figure 3 shows examples of errors that MINS can commit, such as detecting apoptotic nuclei as live cells (asterisks in panels in Figure 3Aa, Ab, Ad), over-segmentation (arrow and arrowheads in Figure 3Ac) or under-segmentation (arrow in Figure 3Ad). Figure 3B shows a sequence of Z-slices of an under-segmentation event, where two cells have been identified as one. Note how MINS segmentation takes the Z-axis into account to segment a volume that comprises all slices where a given cell is present.
Finally, the specifics of the data analysis will depend on the question under study, therefore, guidelines for the analysis process cannot be given here. However, we have found certain initial data transformations to be useful. Figure 4B – D shows plots of the fluorescence values for the markers NANOG and GATA6 in the ICM of the embryo shown in Figure 4A. Figure 4B shows the raw fluorescence values obtained from MINS (after manual correction of under- and over-segmentation). Figure 4C shows the same data after a logarithmic transformation, which separates the values from the plot axes (a square root transformation is also possible and yields a similar plot). Figure 4D shows the same data after automatically correcting the values for the Z-associated attenuation in fluorescence, as explained in step 4.5.2.2 of the protocol. Examples of this Z-associated fluorescence decay are given in the image in Figure 4E and the plots in 4F. Figure 4G shows the effect of the data transformation explained in 4.5.2.2. Colors representing either epiblast (red, NANOG+, GATA6-) or PrE (blue, NANOG-, GATA6+) identity have been added to the plot as a function of the NANOG and GATA6 values. In this particular example, cells where the ratio of log[GATA6]/log[NANOG] >1.25 were considered PrE, cells where the ratio of log[GATA6]/log[NANOG] < 1 were considered EPI, and cells with an intermediate ratio were considered uncommitted (none in this case). All data transformations and plots have been done in Rstudio, however, the choice of software is not critical and will depend on the end user.
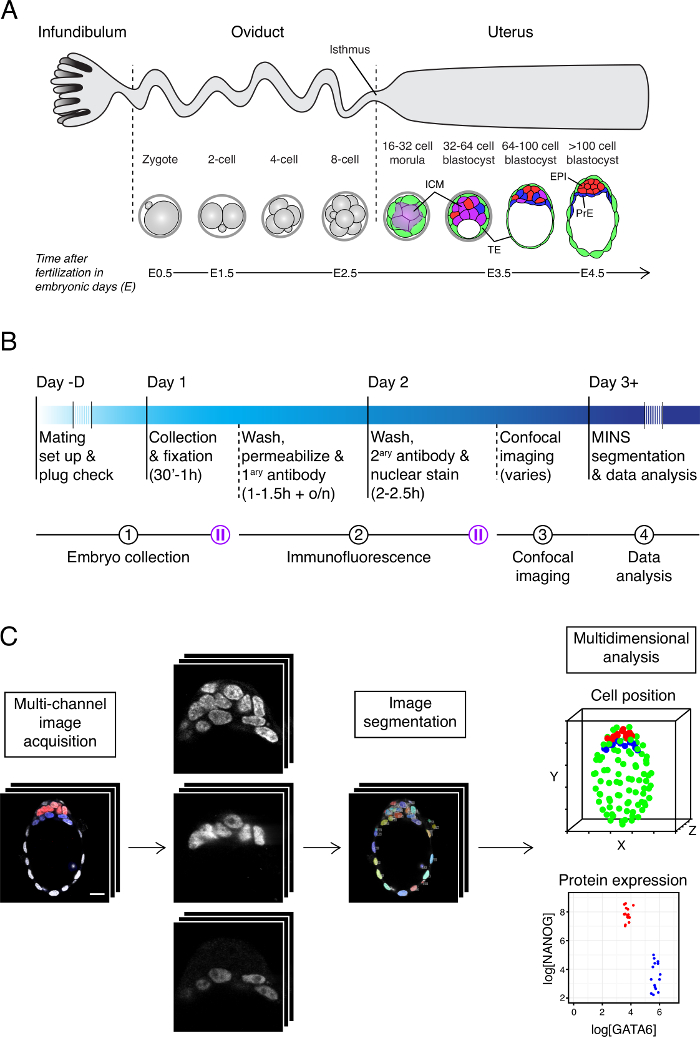
Figure 1. Embryo Location, Timeline and Analysis Pipeline. (A) Schematic of one (of two) mouse uterine horn and the stages of preimplantation development, aligned with the region of the horn where they should be found. (B) Experimental timeline with timing for each step. Steps of the protocol and pause points (blue) are indicated. (C) Summary of image acquisition and analysis pipeline using MINS. Please click here to view a larger version of this figure.
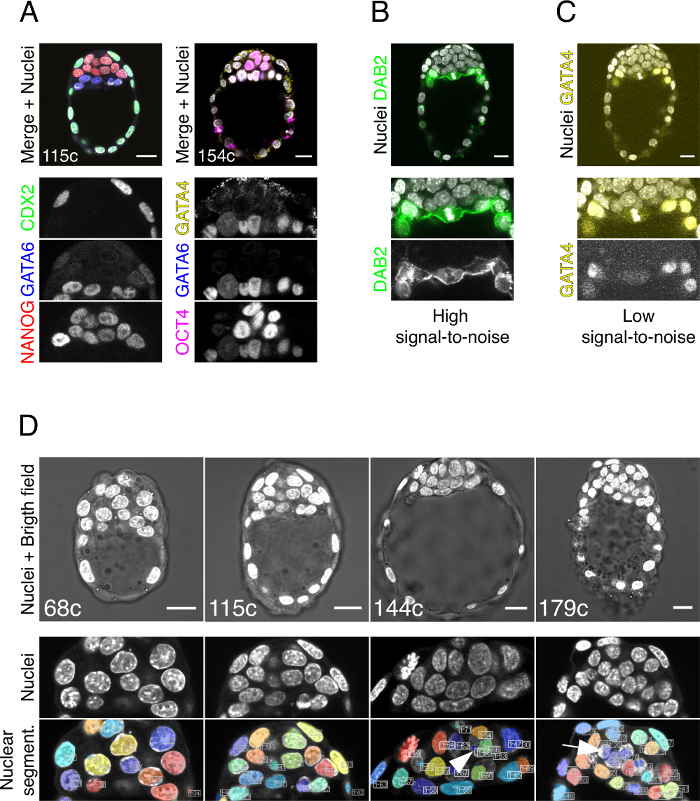
Figure 2. Examples of Immunofluorescence and Nuclear Density. (A) Examples of good immunofluorescence results with tested antibodies for nuclear transcription factors. Anti-CDX2 was detected with an AlexaFluor 488 donkey anti-mouse secondary antibody, anti-GATA6 with an AlexaFluor 568 donkey anti-goat, anti-NANOG with an AlexaFluor 647 donkey anti-rabbit, anti-GATA4 (Santa Cruz, sc-9053) with an AlexaFluor 488 donkey anti-rabbit and anti-OCT4 with an AlexaFluor 647 donkey anti-mouse. All primary antibodies are listed in the Materials Table. Cytoplasmic GATA4 nuclear staining is non-specific, and appears also in Gata4 null embryos (not shown). (B) Example of good immunofluorescence for a cytoplasmic protein, DAB2. It has been detected using an AlexaFluor 488 donkey anti-mouse. (C) Example of bad immunofluorescence, with a low signal-to-noise ratio for a nuclear factor. GATA4 has been detected with an anti-GATA4 raised in goat (Santa Cruz, sc-1237; whereas sc-9053 (rabbit anti-GATA4) was used in (A)) and an AlexaFluor 568 donkey anti-goat. (D) Examples of intact embryos with good nuclear staining showing how nuclear density increases with embryo age and how this increases errors in MINS segmentation (arrowhead – oversegmentation – and arrow – undersegmentation). Scale = 20 µm. Please click here to view a larger version of this figure.
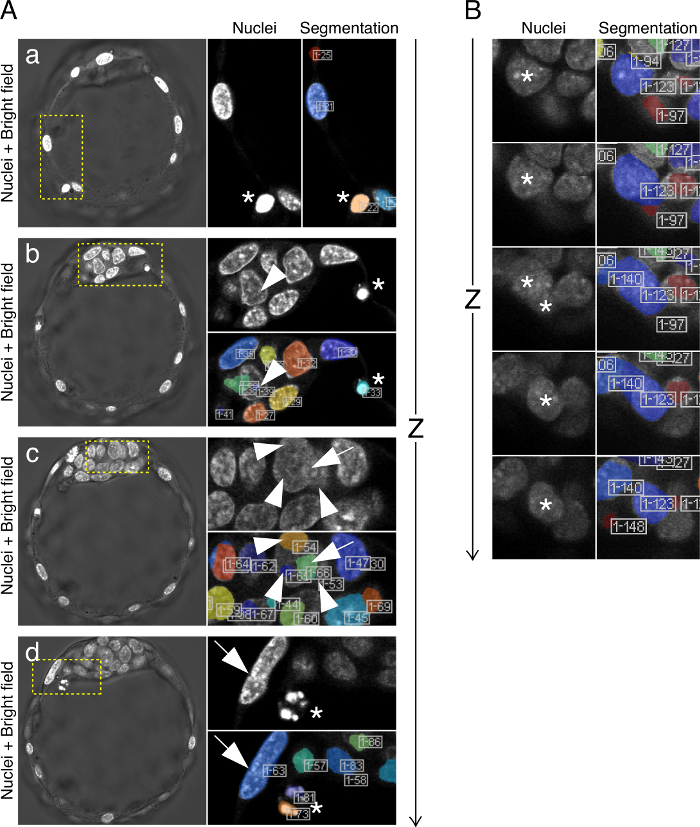
Figure 3. Examples of MINS Errors. (A) Sequence of individual Z-sections from one embryo showing examples of MINS segmentation errors. Apoptotic cells which are nevertheless identified as nuclei are marked with an asterisk (*) in panels a, b and d. In panel b, ID #39 (arrowhead), albeit very small, does identify the corresponding nucleus and can thus be left uncorrected. In panel c, ID #66 (arrow) spans two different nuclei, which have in turn been identified separately (arrowheads, IDs #65 #54 and #53). In panel d, two cells (ID #63) have been identified as a single one (see arrow marking the thin border) and this record should therefore be duplicated for cell counting purposes. (B) MINS detects cells along the Z-axis. The images show a sequence of Z-slices of two cells that have been erroneously scored as a single one (#123). Note how the blue area delimiting the nuclei is continuous along Z. Please click here to view a larger version of this figure.
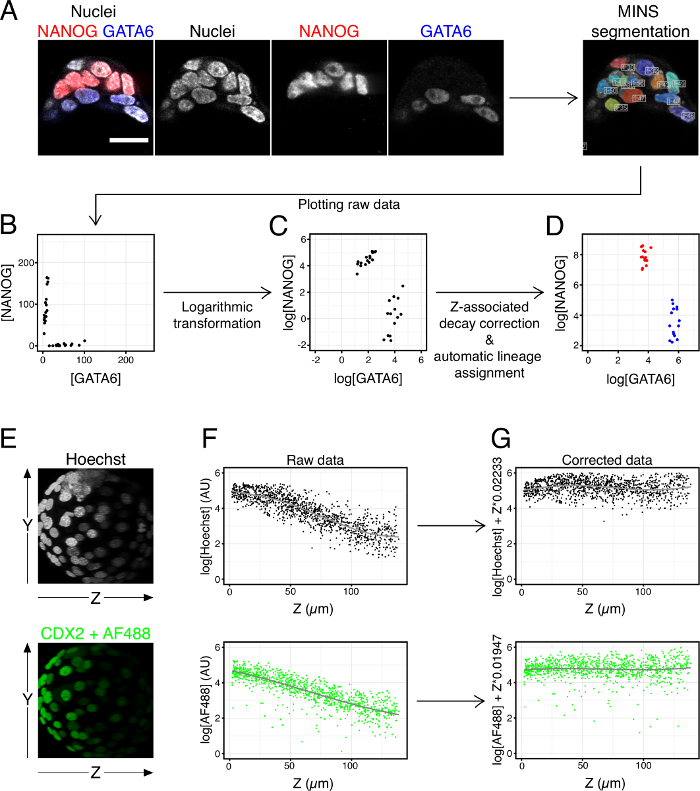
Figure 4. Example of Segmentation and Data Transformations. (A) Images of ICM cells of a blastocyst stained for NANOG and GATA6 and snapshot of nuclear segmentation of the same embryo using MINS on the nuclear staining channel (Hoechst 33342). (B-D) NANOG and GATA6 fluorescence values in ICM cells measured by MINS plotted as raw data (B), after performing logarithmic transformation (C) and after correcting values for the fluorescence decay along the Z axis and automatically assigning cell identities based on the corrected values (D). (E – G) Examples of data correction for fluorescence decay along the Z axis. (E) Images of a blastocyst labeled with Hoechst and anti-CDX2+AlexaFluor 488 (AF488), showing fluorescence decrease along the Z axis. (F) Scatter plots of fluorescence values along the Z axis for Hoechst and AF488 for a pool of embryos including the one shown in (E). Gray line represents the regression curve for each set of values. (G) Same data as in F after compensating fluorescence decay for each cell using the following factor: Z coordinate * the slope of the linear model (see step 4.5.2.2 in the protocol). Panels E – G were originally published by the authors in the Node (http://thenode.biologists.com/99problems/discussion/) Each dot represents one cell. Scale = 20µm. Please click here to view a larger version of this figure.
| User Input |
| Brightness threshold* (facilitates nuclear detection in dim images) |
| Relative X/Y-Z resolution |
| Channel to segment |
| Nuclear diameter |
| Image noise level |
| MINS Output |
| Segmentation Z-stack with nuclei IDs |
| Nuclear volume |
| XYZ coordinates for each nucleus |
| Average and summation of fluorescence values for each fluorescence channel and nucleus |
| Advantages |
| Accurate segmentation (>85%) of nuclei throughout the stack — recognizes all occurrences of a nucleus in adjacent z-sections |
| Single cell resolution |
| Unsupervised segmentation of batches of embryos |
| Segmentation pipelines can either be saved and re-used or adjusted for each particular embryo |
Table 1. MINS Features
| Load Image | At the prompt, introduce the Z relative resolution for the image. It can be obtained from the file metadata using the default reader or applying the following formula: X (or Y) resolution/Z resolution (all in µm). |
| Enter the sequence number (1 – 5) for the channel to be segmented. Using the DNA stain channel for segmentation will detect all cells in the image, however, other channels may be segmented separately too. | |
| Enter the frame to begin segmentation at (1 by default). Only applies to files with multiple time frames. | |
| Enhance Image (optional) | The white and black point of the image can be adjusted to facilitate detection. Generally lowering the white point and re-scaling to 0 – 255 (for 8-bit images) or 0 – 4096 (for 12-bit images) will make the image brighter and facilitate detection of dim nuclei. |
| Detect Nuclei | Select the estimated nuclear diameter (generally between 30 and 40 px in blastocysts). |
| For noisy images the noise level can be adjusted. Otherwise, the default value will be applied. | |
| Segment Nuclei | Use default parameters. |
| Classify Nuclei | MINS can distinguish trophectoderm (TE) from inner cell mass (ICM) cells in preimplantation embryos based on position. This can also be done manually or based on fluorescence levels if a TE marker is used. |
| Export Results | Select the files to be generated: |
| – segmentation overlaid over the channel used (.tiff sequence file), | |
| – segmentation only (.tiff sequence file), | |
| – spreadsheet with IDs for all cells detected, XYZ coordinates and fluorescence levels (average and sum) for all channels for each cell (.csv file). | |
| All files exported by default. Files are saved in the same directory where images are located. |
Table 2. MINS Segmentation Pipeline
