To genetically incorporate a Uaa into a protein in neurons, the first important step is to design appropriate gene constructs to deliver and express genes efficiently in neurons. There are three genetic components for Uaa incorporation: (1) the target gene with the TAG amber stop codon introduced at the chosen site for Uaa incorporation (2) an orthogonal tRNA to recognize the mutated TAG stop codon, and (3) an orthogonal aminoacyl-tRNA synthetase to charge the Uaa onto the orthogonal tRNA. Each component needs to be driven by the appropriate promoter. These three gene cassettes can be included in one plasmid or separated in several plasmids. To express PIRK in neurons, the photo-reactive Uaa Cmn is incorporated into Kir2.1 using the orthogonal tRNALeuCUA/CmnRS (Cmn-tRNA synthetase) pair evolved to be specific for Cmn21,29. Residue Cys169 of Kir2.1 gene is mutated to the TAG amber stop codon (Kir2.1-TAG), and the fluorescent protein mCitrine is fused to the C-terminus of Kir2.1 to visualize PIRK expression in neuronal culture (Figure 1).
Obtaining good-quality neuronal culture is a prerequisite for successful PIRK experiments. Postnatal day 1-4 Sprague Dawley rat pups are generally used for neuronal culture preparation, yet rat embryos are also frequently used. When plated onto coverslips in 24-well plates at 1.0-1.5 x 105 cell/well density, one should see well-separated healthy neurons with not too much microglia or other debris (Figure 2A). Healthy neurons have extensive processes, and cell bodies (soma) are plump (Figure 2B). Distinctively visible suborganelle structure is usually a sign of unhealthy culture. Neuronal culture can be maintained for 2-3 weeks at 35 °C in the 5% CO2: 95% air humidified incubator.
Calcium phosphate (Ca-P) transfection is a very gentle transfection method appropriate for sensitive neuronal culture. However, it requires extreme precision and caution to achieve high transfection efficiency in neurons. Stock solutions need to be stored at 4 °C, and mixed to make 2x BBS buffer on the day of transfection. Precise pH adjustment is key to reproducibly get successful results. The 2.5 M CaCl2 solution should also be made fresh on the day of transfection. All buffers need to be filtered. Moreover, fresh DNA plasmids with high purity and quality improve the results. For best results, one can obtain fresh DNA mini-prepped from not overgrown Escherichia coli culture (~12 hr at 37 °C in the shaking incubator). After all the buffers are ready, bring the solutions in the tissue culture hood and prepare for transfection. Using a vortex at a low speed, agitate the transfection solution while mixing. Immediately add the mixed solution to the neuronal culture, and bring the culture dish back to the incubator. Stop the transfection after 45 min-1 hr, and add the original growth media back to the culture for continuous incubation. The transfected neurons can be visualized from 6 hr after transfection is terminated (Figure 3). In the presence of Cmn in the culture media, PIRK is expressed and PIRK-expressing neurons are green-fluorescent due to the mCitrine protein fused to the C-terminal of PIRK (Figure 3D). PIRK-expressing neurons have normal basal physiology and they are capable of firing action potentials in response to injected currents (Figure 4). However, this series of action potentials is suppressed immediately with a brief light pulse shone to the cell (Figure 4A). When 0.5 mM BaCl2, a Kir2.1 specific inhibitor, is added to the bath, neuronal firing is then resumed, indicating that the previous suppression was due to the activation of Kir2.1 by the light (Figure 4B). Untransfected control neurons do not respond to the light or Ba2+ (Figure 4C, D).
To express PIRK in vivo, highly pure and concentrated DNA plasmids (2-5 µg/µl) should be prepared. Specific plasmids used in these experiments are shown in Figure 5A. In addition to the orthogonal tRNALeuCUA/CmnRS pair and the target Kir2.1-TAG gene, a gene coding green fluorescent protein with a TAG mutation (GFP-TAG) is also co-electroporated as a fluorescent reporter. Detection of GFP fluorescence would indicate the successful delivery of all three plasmids, since TAG suppression in GFP would require both the tRNALeuCUA and the CmnRS; Cmn incorporation in GFP-TAG would suggest Cmn incorporation in Kir2.1-TAGas well, because both genes are present in the same cell. The three gene constructs are all injected into the lateral ventricle of mouse embryos on E14.5 (Figure 5B, left panel). After electroporation, return the embryos to the abdominal cavity to allow the embryos to continue development. Two days later, inject about 2-5 µl Cmn-Ala (500 mM) to the electroporated side or both sides of the lateral ventricle, in the similar manner as DNA injection (Figure 5B, middle panel). Again, return the embryos to the abdominal cavity for continuous development. The embryos can be harvested on E17.5 for imaging or electrophysiological assays. As expected, only with Cmn-Ala injection, green fluorescent cells are observed in the mouse neocortex. Cells with both red and green fluorescence should have Cmn incorporated into Kir2.1-TAG to make PIRK channels (Figure 6A). Whole-cell recording from the embryonic neocortical slice is done similarly as it is done on neuronal culture. The green and red fluorescent neurons have no inward current at negative holding potential, but a brief pulse of light rapidly activates the inward current (Figure 6B). The current is completely blocked by adding Ba2+, confirming that it is generated by PIRK.
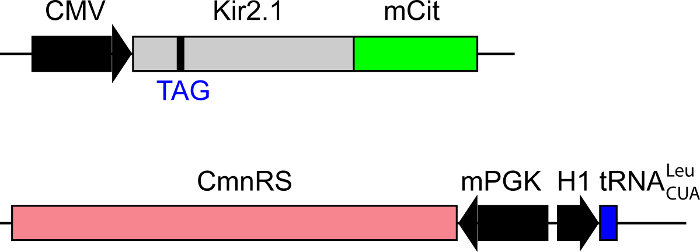
Figure 1: PIRK expression plasmid set for neuron culture. Scheme showing exemplary plasmid design for PIRK expression in neuronal culture. Top plasmid encodes Kir2.1 gene (grey) with a single amino acid mutation in C169 site under cytomegalovirus (CMV) promoter. C169 is mutated to the TAG amber stop codon, where the Uaa Cmn would be incorporated. Kir2.1 gene is followed by mCitrine gene (green). mCitrine is fused to the C-terminal of the Kir2.1 gene to visualize PIRK expressing cells. Bottom plasmid encodes CmnRS (pink), the Cmn specific synthetase, driven by the mouse phosphoglycerate kinase-1 (mPGK) promoter. tRNALeuCUA (blue), the orthogonal tRNA21, is also expressed in the same plasmid driven by the H1 promoter, but in a reverse direction.
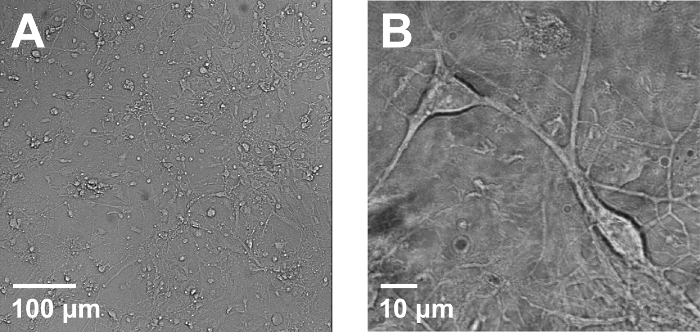
Figure 2: Rat hippocampal primary neuronal culture. Exemplary pictures showing healthy rat hippocampal neuronal cultures. (A) A DIC image of neuronal culture on 10 days in vitro (DIV). Neurons mature as they branch out dendrites and axons. Microglia cells (small and round cells without processes) coexist but do not overwhelm the culture. (B) A magnified DIC image of healthy cultured neurons. A healthy neuron exhibits plump cell body (soma) and pronounced dendrites/axons. Please click here to view a larger version of this figure.
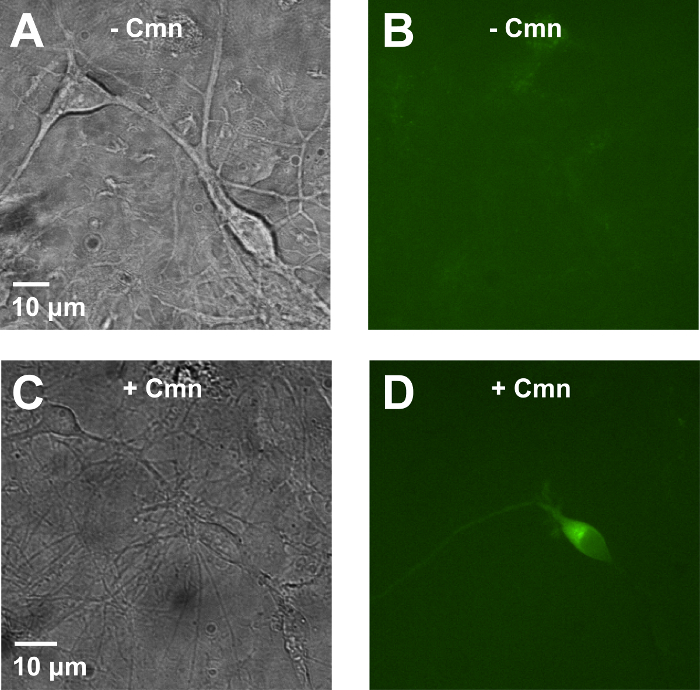
Figure 3: PIRK expression in cultured primary neurons. DIC and fluorescence images of rat hippocampal neurons cultured in vitro and transfected with gene constructs depicted in Figure 1 for PIRK expression. When Cmn is not added in the culture after transfection (A and B), no green fluorescence is detected from the neurons. In the presence of Cmn (1 mM) in the growth media, transfected neurons are capable of expressing full-length PIRK-mCitrine proteins, thus showing green fluorescence (C and D). Please click here to view a larger version of this figure.
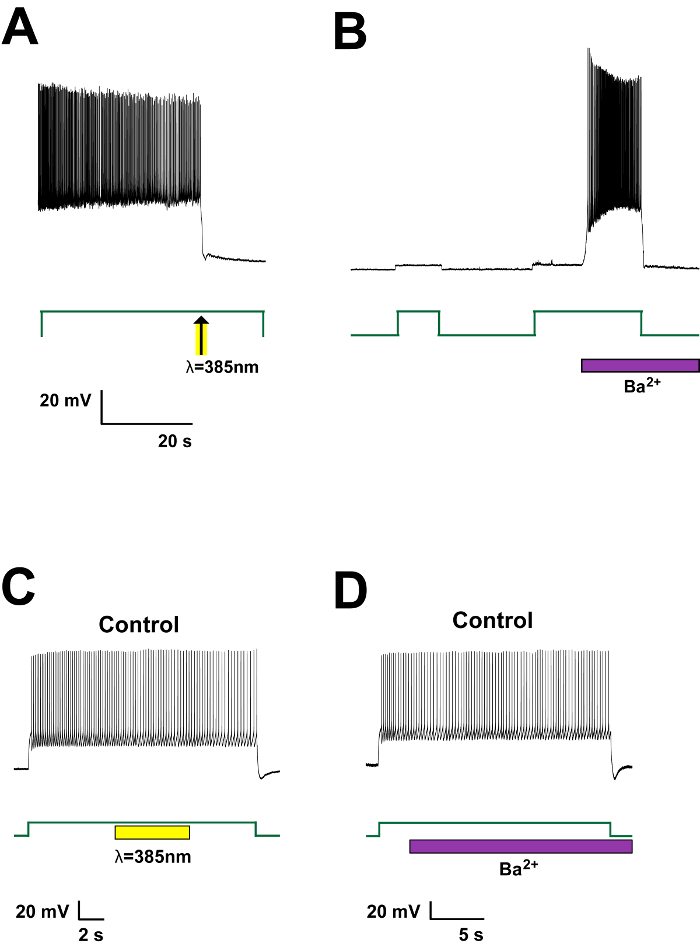
Figure 4: Light activation of PIRK suppresses firing from rat hippocampal neurons. (A) A single light pulse suppresses activity of the hippocampal neuron expressing PIRK. Representative voltage traces are recorded continuously in current-clamp. Action potential firing is evoked by 20 pA current injection (I-step). Light exposure (385 nm, 40 mW/cm2, 1 sec; indicated with arrow) rapidly and completely suppresses neuronal firing. Firing is restored with extracellular 500 mM BaCl2, which selectively inhibits Kir2.1 channels. (B) Ultraviolet (UV) illumination (385 nm, 40 mW/cm2, 6 sec; indicated with yellow box) does not alter excitability of control, untransfected neurons. Action potential firing is evoked by 50 pA current injection (I-step). (C and D) Neither UV illumination nor BaCl2 (500 mM) affects excitability of control neurons. Action potential is evoked by 50 pA current injection (I-step).
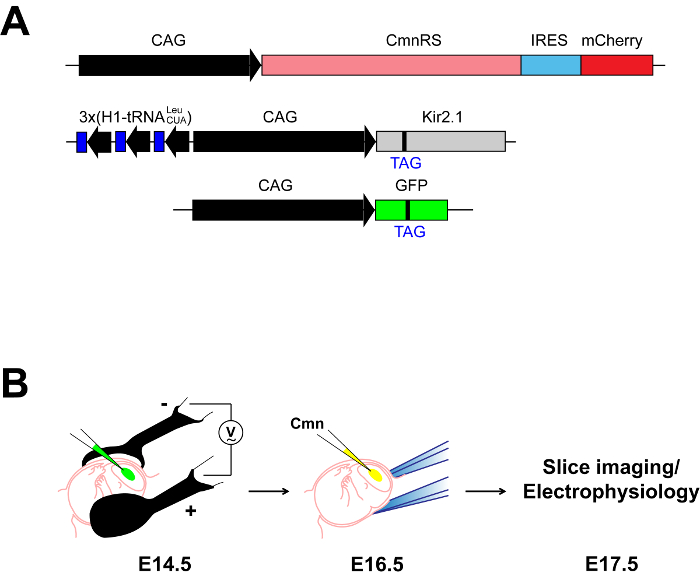
Figure 5: Procedures for PIRK expression in the mouse neocortex in vivo. (A) PIRK expression plasmid set. Top plasmid: the plasmid for CmnRS driven by the CAG promoter and mCherry via internal ribosome entry site (IRES). mCherry fluorescence would verify successful expression of CmnRS. Middle plasmid: the plasmid for Kir2.1 gene coupled with three copies of tRNALeuCUA, the orthogonal tRNA for Cmn incorporation21. Bottom plasmid: the plasmid for GFP_Y182TAG. The gene for GFP is engineered with an amber stop codon at the permissive Tyr182 site (GFP_Y182TAG) to visualize successful incorporation of Cmn to TAG sites21. (B) Cartoon showing an experimental procedure for PIRK expression in vivo. Gene constructs for PIRK expression are injected into the mouse neocortex (E14.5) and electroporated in utero (left panel). Two days later, Cmn-Ala is injected to the ventricle in the electroporated side or both sides of cerebral hemispheres (middle panel). Slice imaging and electrophysiological assay can be performed on E17.5-E18.5 (right panel).
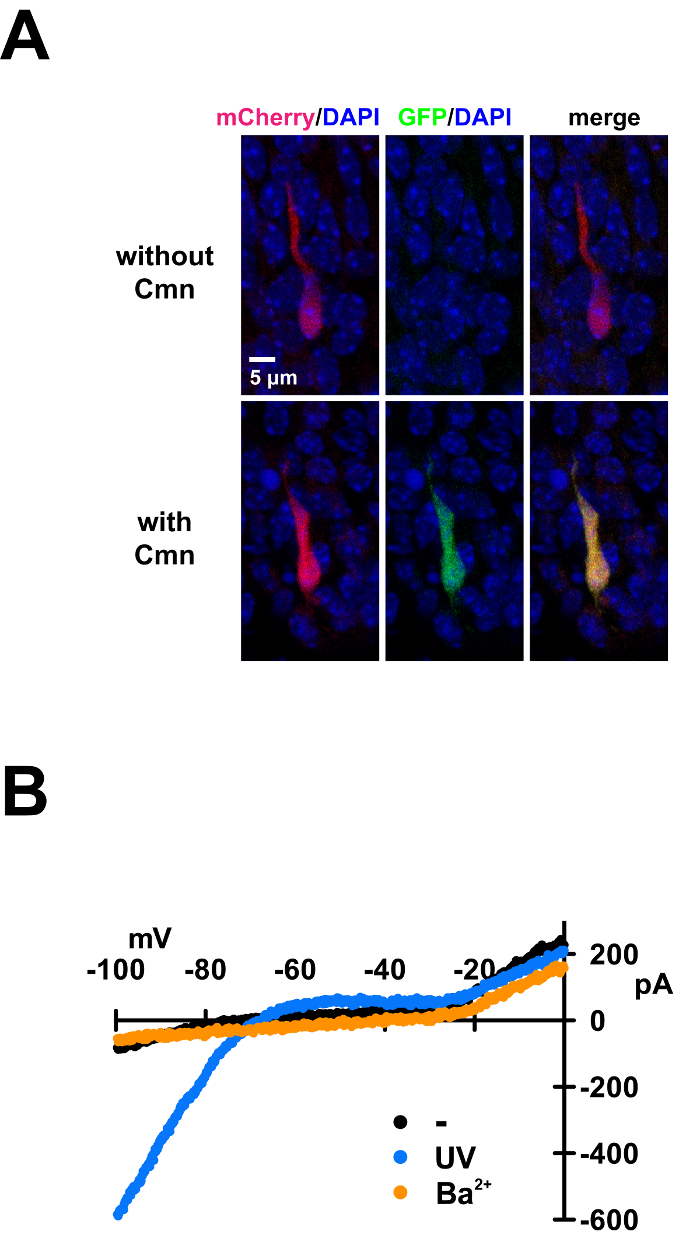
Figure 6: PIRK expression in the mouse neocortex and light activation. (A) Fluorescence images of mouse embryonic cortical neurons showing the incorporation of Cmn into GFP and Kir2.1 proteins in vivo. The three gene constructs in Figure 5A are electroporated in utero. mCherry fluorescence demonstrates successful expression of the Cmn specific synthetase gene, CmnRS. GFP fluorescence is detected only with Cmn-Ala injection (bottom), indicating Cmn incorporation in GFPTAG and likely Cmn incorporation in Kir2.1TAG. Thus, green and red fluorescence indicates successful expression of all three plasmids and Cmn incorporation. (B) I-V plot of currents recorded from mice neocortical neurons showing light-dependent activation of PIRK. Two days after gene constructs in Figure 5A were electroporated, Cmn-Ala was injected in utero; 12-48 hr after Cmn-Ala injection, neocortical acute slices were prepared from the embryos. PIRK-expressing neurons in the slices, detected by both red and green fluorescence, are recorded before (black) and after (blue) light exposure (385 nm, 8 mW/cm2, 10 sec for saturated exposure). BaCl2 (500 mM) is added to verify PIRK-specific currents after photoactivation (orange).
