1. Sample Preparation
- Assemble samples for short chain carboxylic acid (SCCA) extraction. Prepare 1.0 g of coffee seed at a time to ensure that enough sample will remain after processing.
- If samples were frozen prior to the grinding process, keep the tissue frozen throughout processing to prevent freeze/thaw damage and sample oxidation. Remove the sample from the freezer or sub-zero storage only as needed for grinding.
- Flash freeze fresh samples in liquid nitrogen immediately prior to sample grinding. To minimize sample handling, flash freeze samples by placing them in a mortar pre-filled with liquid nitrogen.
- Analyze liquid samples immediately following generation, or flash freeze in liquid nitrogen and store at -20 ºC or -80 ºC until analysis. Prior to analysis, remove frozen samples from storage and allow to thaw. For liquid samples proceed to step (3.5) for processing.
- Wear appropriate personal protective equipment (including safety glasses, gloves, and lab coat) before working with liquid nitrogen.
- Triple-grind tissue samples to a uniformly fine powder (i.e., of uniform particle size) in liquid nitrogen using a ceramic mortar and pestle.
NOTE: Attaining a uniform particle size is essential for maximizing SCCA extraction efficiency.- Pre-chill the mortar and pestle with liquid nitrogen prior to the addition of the sample. Keep the mortar filled with a small volume of liquid nitrogen as the sample is added.
- Using a ladle, add enough liquid nitrogen to the mortar to completely submerge the sample.
- Add easily ground samples, such as leaves or roasted coffee, to the liquid nitrogen and crush using a circular grinding motion. Begin to grind samples when the liquid nitrogen level has decreased to the point where it just barely covers the samples.
- Add hard to grind samples, such as raw coffee seeds, to liquid nitrogen and allow them to freeze for 10-30 sec (or until the liquid nitrogen stops vigorously boiling) before grinding. Break the tissue into smaller fragments using a vertical crushing motion, and then complete tissue grinding using a circular grinding motion.
- Repeat steps 1.3.2-1.3.4 twice more, so that samples are ground a total of three times. Typically, three successive rounds of grinding will reduce samples to a powder with a flour-like consistency.
- If a flour-like consistency is not achieved, repeat steps 1.3.2-1.3.4 until samples are reduced to a powder of uniformly small particle size (extraction efficiency is inversely proportional to particle size).
- Transfer powder to glass vials or 1.5 ml microcentrifuge tubes. Initiate downstream processing immediately after grinding (recommended), or store samples at -80 ºC until samples are ready for extraction.
- If samples must be stored before analysis, split the sample into 500 mg aliquots (or 500 µl aliquots for liquid samples) and split among multiple tubes for storage. Avoid subjecting samples to repeated freeze/thaw cycles, as these may alter sample composition and negatively impact future sample measurements.
2. Organic Acid Standard Preparation
- Assemble authentic standards for the SCCAs of interest. These will be used to create external and internal standard solutions for use in determining SCCA concentrations. For coffee samples include citric, malic, acetic, and lactic acid as acids of interest and adipic acid as the internal standard.
- Ensure sure that the internal standard selected is not found naturally in the sample, and that the standard does not co-elute with other peaks in the sample profile.
- Run standard curves for each SCCA of interest, and determine the linear response range for each SCCA (see section 4 for running instructions). Make sure to run standard curves for each SCCA to be measured in the sample.
NOTE: Standard curves may be run in either buffer or the background of the sample to be assayed. In the second case ("standard addition"), values for the peak area of each standard curve point will be determined by subtracting away the background of the sample. - Pre-label all tubes and glassware needed with the SCCA acid name, concentration, and the date of the assay.
- Prepare stock solutions for each standard at a known concentration (10 mg/ml) using a volumetric flask to ensure accuracy during standard preparation.
- Dissolve solid SCCA standards (citric acid and malic acid) in ultrapure water (18.2 MΩ) to achieve the concentration desired for the stock solution.
- Create a 10 mg/ml stock solution by adding 100 mg of standard to a 10 ml volumetric flask. Fill the volumetric flask to the 10 ml line with ultrapure water to dissolve the acid.
- Create a lower concentration of acid standard or gently heat the flasks to drive difficult-to-dissolve SCCA standards, like adipic acid, into solution.
- Dilute liquid phase acid standards (acetic acid and lactic acid) in ultrapure water to achieve the desired concentration.
- Pre-fill a volumetric flask with 5 ml of ultrapure water. Add enough acid to prepare a 10 mg/ml solution (calculated using the acid density provided by the vendor) to the flask, and then add enough water to bring the final volume of the solution to 10 ml.
- Dissolve solid SCCA standards (citric acid and malic acid) in ultrapure water (18.2 MΩ) to achieve the concentration desired for the stock solution.
- Transfer each stock solution to a clean 15 ml glass tube, with a polytetrafluoroethylene (PTFE) lined cap, and seal with plastic paraffin film. Stock solutions can be stored in sealed tubes at 4 ºC for 1 week.
- Ensure that SCCA standards have not precipitated out of solution prior to use if stock solutions have been refrigerated after preparation. Drive precipitates back into solution through gentle heating.
- Prepare the SCCA extraction solution. Include the internal standard at a concentration sufficient for detection in the samples (0.05 mg/ml). Prepare enough solution to extract all samples.
- Prepare 50 ml of SCCA extraction solution by diluting the internal standard stock solution to 0.05 mg/ml in ultrapure water. Add 250 µl of the internal standard stock solution (10 mg/ml) to 49.75 ml of water.
- If the extract needs to be diluted, adjust the internal standard concentration in the extraction solution so that the final concentration will fall within the limits of quantification (giving a detectable, but not over-saturated, peak, see 5.2.2 below) of the CE system after dilution (0.05 mg/ml).
- Prepare fresh extraction solution for each series of extractions (i.e., for each experimental series).
- Prepare standard curve samples (a series of appropriate dilutions) for SCCAs of interest, using a minimum of at least 5 points. The standard curve concentrations employed will need to be in the linear response range for a given SCCA, and span the SCCA concentrations expected in the samples.
- Include the selected internal standard in the standard curve solutions to allow quantification of the internal standard in samples. The internal standard will be used to aid in peak identification and calculating extraction efficiency (section 6).
- Dilute each SCCA to the concentrations determined above (0.01, 0.02, 0.04, 0.06, 0.08 mg/ml; see above, 2.4.2) in new microcentrifuge tubes (one for each concentration/standard curve point) using ultrapure water. Prepare 1 ml of solution for each standard curve concentration point. Ensure that each concentration point contains all four acid standards and the internal standard at the correct concentration.
- Prepare new standard curve points for each set of samples to be run on the capillary electrophoresis system. Hold the standard curve SCCA samples at 4 ºC until analysis, which will occur following the extraction of SCCAs from target tissues (section 3).
3. Organic Acid Extraction
- Pre-label sample tubes, preparing at least one tube for each sample to be extracted.
- Remove samples to be extracted from storage and place them on ice while weighing out material for extraction.
- Weigh out 100 mg of sample for SCCA extraction.
- After weighing each sample, transfer tissue to a clean 1.5 ml microcentrifuge tube. Measure out an amount of sample as close to the target mass as possible to reduce variability.
- Keep track of the mass of each sample measured, as the amounts of SCCAs detected will be normalized using the sample mass (see 6.5.2, below).
- After weighing all samples, add 1 ml of extraction solution (the water + internal standard mixture from step 2.4) to each of the sample tubes. Keep the ratio of extraction solution to tissue mass the same across all extractions (in this case 1 ml for 100 ± 5 mg tissue). Mix well by vortex for 10 sec.
- Allow samples to sit at room temperature for 1 hr. During this hour, mix each tube every 15 min as in step 3.4.
- After 1 hr of extraction, mix the samples one final time and transfer sample tubes to a microcentrifuge. Centrifuge samples at 4 °C, 10,000 x g for 10 min to precipitate solid material (cell walls, particulate matter, etc.).
- When processing liquid samples, add the appropriate concentration of internal standard, mix briefly and centrifuge. After centrifugation, treat liquid samples identically to the extract from solid samples.
- Check the pH of the samples to confirm that they are compatible with the pH range of the running buffer in the detection kit (step 4.6) or buffer system being employed. As the volumes of sample are usually quite small, pH can be monitored using pH paper.
- Prepare to filter samples using syringe-mounted disc filters (3.8).
NOTE: Prevent sample loss by ensuring that each filter is correctly attached to a syringe before transferring supernatant. Prepare one syringe equipped with a disc filter for each sample to be analyzed (including standard curve samples). - After centrifugation of samples and preparation of the syringe filters, transfer the supernatant from each sample to a 3 ml syringe equipped with a 0.2 µm syringe filter. Use a new filter and syringe for each sample. Filter all samples, including standard curve samples and buffer controls, prior to running on the CE system.
- Filter the sample directly into a clean microcentrifuge tube. After filtering, close the tube containing filtrate and discard the syringe/filter apparatus.
- Immediately place filtered samples into the CE system for SCCA detection; or store samples at 4 °C overnight. If samples are to be stored overnight, seal the tubes with plastic paraffin film to prevent gas exchange and sample evaporation.
- If samples must be stored longer than a day after filtering, flash freeze extracts in liquid nitrogen or freeze at -20 ºC. After thawing, however, ensure that each sample is examined for precipitate formation and filter if necessary (as in step 3.6).
4. Setting Up the SCCA Detection Run
- Consult the CE user manual for programming- and control software-specific details.
- Prepare capillary electrophoresis (CE) sample vials for SCCA detection. Ensure vials are properly labeled, clean, and free of defects.
- Wash and dry the caps of the CE vials before use.
- Wash caps by submerging them in ultrapure water and allowing them to soak overnight. After soaking the caps, discard the soaking solution and rinse the caps twice more with ultrapure water.
- Transfer rinsed caps to a clean drying surface lined with lint-free tissue and allow them to air-dry. Ensure caps are completely dry before use to avoid pressure failures.
- Transfer 1 ml of sample to each CE vial, being careful to avoid accidentally splashing the sample into the neck of the vial. After transfer, place a CE cap on each vial.
- If a dilution is needed, dilute the coffee seed samples 1:10 or 1:100 directly in the CE vial using ultrapure water. For dilutions greater than 1:100, create an intermediate dilution to avoid pipetting errors.
- Prepare one vial for each standard curve point by transferring standard curve solutions (1.0 ml) from step 2.5 to CE vials. Cap each tube after transfer.
- Prepare a 0.1 M solution of sodium hydroxide (NaOH) by adding 0.04 g of NaOH to a beaker containing 90 ml ultrapure water and allowing the pellets to dissolve. Transfer the 0.1 M NaOH solution to a 100 ml volumetric flask and bring the total volume to 100 ml.
- Add 1 ml of 0.1 M NaOH solution to a clean CE vial and cap.
- Prepare CE buffer vials for each batch of samples to be run on the CE system. Separation kits are available or custom buffers can be used13.
- Prepare 1 vial with 1 ml of starting buffer solution and cap.
- Prepare three vials with 1 ml each of running buffer solution and cap. Replace the running buffer before each batch of samples and standard curve points, or after 35 samples, whichever occurs first.
- Fill three additional vials with 1 ml of ultrapure water and cap each vial.
- Prepare 1 empty "waste" vial with cap.
- After preparing the vials described in step 4.8, load the vials containing the buffers and water, as well as the waste vials (from steps 4.8.2-5) into the buffer tray of the capillary electrophoresis system, as per manufacturer instructions15. Carefully note the position of each vial for auto-sampler programming.
- Load the vials containing the standard curve points and the vials containing the samples to be assayed into the inlet sample tray, as described in the manufacturer's instructions. Note the position of each vial for auto-sampler programming (see steps 4.11.1 below).
- Prepare conditioning and sample separation methods (programs for these are provided in Tables 1 and 2, respectively), and write a sample sequence file as per instrument operating instructions. Use the separation method detailed in Table 2: rinse the column with initiator (20 psi) for 0.2 min; rinse with accelerator (20 psi) for 0.5 min, inject samples (0.5 psi) onto the column for 0.09 min, separate samples at 20 kV for 12 min, rinse with NaOH (20 psi) for 0.5 min, and finally rinse with water (20 psi) for 0.5 min.
- Using the CE control software, write the sample running sequence (i.e., the worklist; a list file detailing the order in which the samples will be analyzed, and the method to be used to separate each sample) using the "sequence" spreadsheet interface. Each row on the spreadsheet will correspond to a sample run and produce a single data file.
- Ensure that when writing the sequence file, samples are identified using the correct position in the sampling tray. Ensure that each data file has a unique name to prevent the software from overwriting previous files. Program CE capillary conditioning runs as a separate sequence prior to the sequence run containing the sample separation method.
- Begin the sample sequence run with the standard curve solutions, followed by SCCA samples, and end with a second run of the standard curve solutions. This will allow a calculation of the degree of any signal loss occurring during sample analyses.
- Using the CE control software, write the sample running sequence (i.e., the worklist; a list file detailing the order in which the samples will be analyzed, and the method to be used to separate each sample) using the "sequence" spreadsheet interface. Each row on the spreadsheet will correspond to a sample run and produce a single data file.
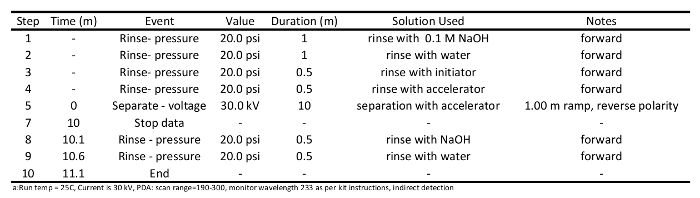
Table 1: Conditioning method program used to prepare the capillary for short chain carboxylic acid separation via capillary electrophoresisa.
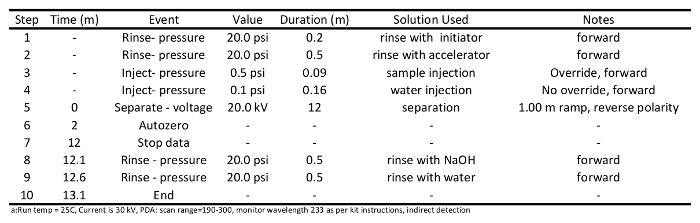
Table 2: Separation method program used to analyses short chain carboxylic acids via capillary electrophoresisa.
5. SCCA Detection Run Execution and Data Collection
- Initiate capillary conditioning runs. Expect to condition the capillary two or three times before the column is ready for sample separation. Column conditioning should be performed as described in Table 1. Briefly, rinse column with 0.1 M NaOH (20 psi) for 1 min, rinse with water (20 psi) for 1 min, rinse with initiator (20 psi) for 0.5 min, rinsed with accelerator (20 psi) for 0.5 min, separate accelerator at 30 kV for 10 min, rinse column with 0.1 M NaOH (20 psi) for 0.5 min, and finally rinse the column with water (20 psi) for 0.5 min.
- Condition the capillary prior to each sample sequence run. Achieve proper conditioning by keeping the separation voltage is constant throughout the run and observing a flat baseline on the photodiode array trace.
- After conditioning, initiate sample separation by opening the "control" menu in the software and selecting (i.e., clicking on) "run sequence." Monitor the photodiode array (PDA) trace to ensure proper separation.
- Observe the first trace and ensure that it has a flat baseline and cleanly resolved (baseline resolution), individual acid peaks. Over-loaded samples will show long peak tails, or peak tailing, and will need to be diluted (Figure 1).
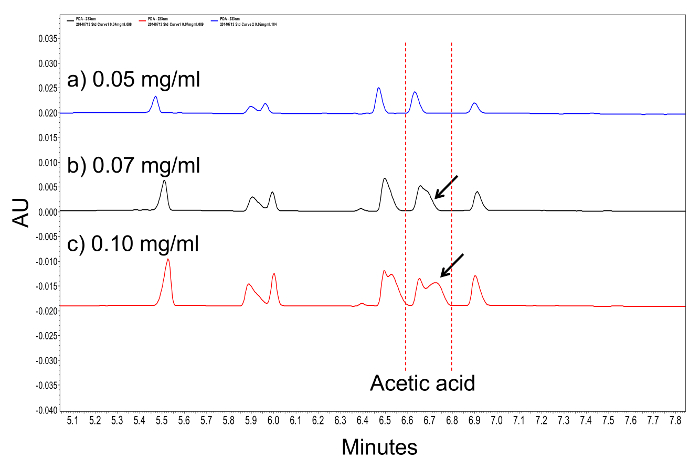
Figure 1: A comparison of PDA traces highlighting an overloaded sample. As analyte concentration increases, individual peak geometry may begin to become asymmetric. At (a) 0.05 mg/ml, acetic acid presents a well-defined, bilaterally symmetrical peak. As the concentration of acetic acid increases to (b) 0.07 mg/ml and (c) 0.10 mg/ml, a peak tail forms (arrows). This peak tailing is a good indication that the sample is overloaded. Please click here to view a larger version of this figure.
- At the conclusion of the sequence run, open the data files one at a time in the CE analysis software and integrate the peaks.
- Integrate sample peaks.
- Open the first file and set the automatic integration steps to exclude the first 3 min of the run (where the voltage, and therefore the direction of column flow, is inverted as described in Table 1). In the same menu, set peak selection criteria to a minimum peak width of 50 units and peak area of 100 units (or the manufacturer's default settings) to provide a moderately high level of selectivity, separating acid peaks from background noise in the trace.
- Manually check peak boundaries and baselines for each PDA trace to ensure proper peak integration. Improperly integrated peaks (i.e., a slightly asymmetrical SSCA peak that has been integrated as two separate peaks by the automated software) will need to be re-integrated manually.
- Integrate sample peaks.
- After integration, view the percent area report, then highlight and copy the percent area report and paste it into a separate spreadsheet. Repeat for each sample to construct the full run peak report with each row representing a single peak (i.e., each peak should contain a single compound if the separation is working well).
6. Data Analysis
- Prepare data for analysis using the spreadsheet constructed in (5.4). Begin analysis by labeling the acid standards for each of the standard curve points, including the internal standard.
- Calculate a retention index for each peak by dividing the retention time for each peak in the sample by the retention time of the internal standard in that sample. Sort the data set by the resulting retention index to identify the peaks of interest in each sample.
- After identifying the peak for each acid of interest, construct standard curves for each acid using the external standard curve points.
- Generate a standard (concentration) curve for each SCCA standard by determining the peak area for each concentration of the SCCA standards, and plotting concentration on the x-axis vs. peak area on the y-axis. Plot the linear regression for each SCCA standard curve and define the slope equation (y = mx + b).
- Ensure that the R2 values of the linear regressions are 0.90 or higher as the quantifications are most accurately performed in the linear range of the standard curve.
- Calculate the acid concentration for a given SCCA in a sample using the peak area and the slope equation for the linear regression line of the standard curve (calculated in 6.3.2 above). Briefly, divide the observed peak area for a given acid by the slope of the regression line for that acid's standard curve.
- Correct the raw concentration values calculated in step 6.4 for sample loss during processing using the internal standard (step 6.5).
- Calculate the correction factor for each sample using the internal standard.
- Divide the actual concentration of the internal standard (i.e., the known amount added at the beginning of the experiment) by the observed value of the internal standard in the sample (i.e., the amount calculated using the slope equation of the standard curve for the internal standard). Multiply the raw concentration values of each SCCA in the sample by this correction factor.
- Divide the internal standard corrected SCCA concentration by the mass of the sample used for extraction to correct for any variation in sample mass. This calculation yields amount of analyte per mass of sample (mg SCCA per mg ground coffee in this example), which can then be converted to the appropriate units for the given study (mg/mg, mg/g, g/g, etc.).
- After calculating SCCA concentrations (normalized to either the mass [for solid or liquid samples] or volume [for liquid samples]), use these values for statistical analysis as per the demands of the experimental design and questions of interest.
This protocol has been successfully utilized to measure the effects of seed treatments on the SCCA content of green coffee seeds. In this experiment, the six treatments were: a saturated microbial suspension of Leuconostoc pseudomesenteroides strain GCP674 in its growth medium (1), an aqueous suspension of GCP674 microbes in water (2), an aqueous solution of acetic and lactic acids (0.15 and 0.4 mg/ml respectively) (3), a spent M1 growth medium treatment (4), dH2O water (5), and an un-treated control (no substance added to seeds) (6). Treatments were applied and allowed to ferment for 24 hours. Four independent replicates were assessed for each treatment. Analysis was conducted for citric (C), malic (M), acetic (A) and lactic (L) acid levels using adipic acid as an internal standard.
For each SCCA and the internal standard, standard curves spanning the range of concentrations predicted to be in coffee samples (5, 10, 20, 40, 60, and 80 ng/µl) were generated. As expected, each acid exhibited a linear response for this concentration range with R-squared values of 0.9876 for citric acid, 0.9987 for malic acid, 0.9998 for acetic acid, and 0.9999 for lactic acid. The internal standard (adipic acid) also exhibited a linear response over this concentration range, yielding an R-squared value of 0.9984. Prior to sample analyses, limits of detection (LOD) and limits of quantitation (LOQ) were determined for each SCCA and the internal standard (adipic acid). The limit of detection of citric, malic, acetic, and adipic acid was 1 ng/µl; and the LOD for lactic acid was 2 ng/µl. The limit of quantitation of citric, adipic, acetic, and lactic acid was 4 ng/µl; and the LOQ for malic acid was 2 ng/µl. To calculate the percent recovery SCCAs and adipic acid, coffee was spiked with individual SCCAs or adipic acid and processed using the protocol described above. The percent recovery was then calculated by using the following formula ([{peak area spiked sample – peak area control sample}/theoretical peak area of calculated concentration of spiked sample {generated by analyzing buffer + SCCA/adipic acid spike}] x 100). Using this method, we calculated percent recoveries of 106.08% ± 12.66 for citric acid, 98.35% ± 1.15 for malic acid, 91.94% ± 3.07 for acetic acid, 97.42% ± 1.48 for lactic acid, and 100.19% ± 2.57 for adipic acid.
To determine the precision of the method, the intra- and inter-day variability of measurements made using CZE was also calculated. To determine intra-day variability, samples of each SCCA and adipic acid were measured from a single coffee sample five times across a 24-hour period, and coefficients of variability (COV; calculated by dividing the standard deviation in peak area [or concentration] for each SCCA by the average peak area [or concentration] for that SCCA, and then multiplying the result by 100) for each compound were calculated using the peak area. To assess the importance of correcting samples using the internal standard, the coefficient of variance for each SCCA was also calculated using the concentrations of each SCCA calculated using adipic acid as an internal standard. As expected, the CZE method was able to precisely measure SCCAs and adipic acid, with COVs of 3.02% for citric acid, 2.64% for malic acid, 3.74% for acetic acid, and 2.01% for adipic acid (lactic acid was below LOD/LOQ for all coffee samples measured). These measurements were repeated across a five-day period, and the CVs ranged from 2.11-5.25% for citric acid, 2.01-5.32% for malic acid, 1.72-3.74% for acetic acid, and 1.26-3.82% for adipic acid. When peak areas were corrected using the internal standard and calculated concentrations of SCCA were used to calculate COVs, the intra-day COVs ranged from 1.08-4.17% for citric acid, 1.47-3.40% for malic acid, and 2.68-5.10% for acetic acid. To assess inter-day variability, SCCAs in a single coffee sample were measured once per day across a five-day period. This experiment was then repeated five times for each SCCA and adipic acid. The COV for each SCCA was then calculated by dividing the standard deviation in peak area (or concentration) for each SCCA by the average peak area (or concentration) for that SCCA and multiplying by 100. To assess the importance of correcting samples using the internal standard, COVs were also calculated using the concentrations of each SCCA calculated using adipic acid as an internal standard. The CZE method was able to reliably reproduce SCCA measurements, with COVs ranging from 5.24-10.02% for citric acid, 6.55-9.47% for malic acid, 7.67-8.63% for acetic acid, and 3.08-6.57% for adipic acid. When peak areas were corrected using the internal standard and calculated concentrations of SCCA were used to calculated COVs, the inter-day COVs ranged from 4.56-6.23% for citric acid, 3.39-4.99% for malic acid, and 9.5-10.94% for acetic acid.
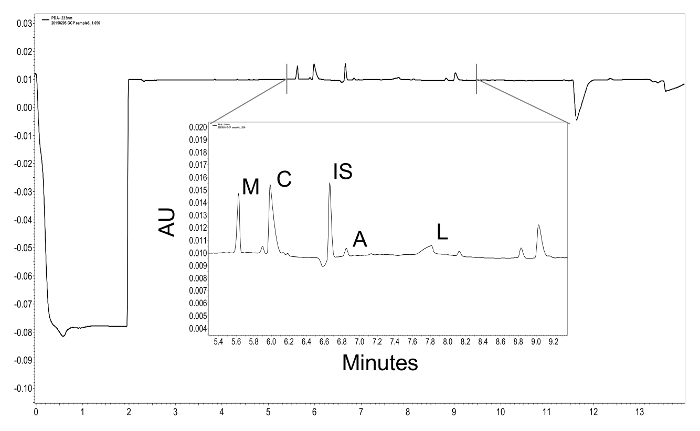
Figure 2: Example PDA traces with peaks indicated. Green coffee samples were diluted 1:10 before loading into CE vials. Acids of interest are indicated: acetic acid (A), citric acid (C), lactic acid (L), malic acid (M) and the internal standard, adipic acid (IS). Please click here to view a larger version of this figure.
Statistical analysis was conducted on corrected acid concentrations using a general linear model (GLM) to determine whether or not treatments altered organic acid levels. A model incorporating "treatment" x "run" (coded as a random factor) was implemented for the GLM. Tukey pairwise comparisons at 95% confidence were used to determine the directionality of changes.
Treatment altered SCCA concentrations in the green coffee. Acetic acid was significantly enriched in all treatments over the no treatment control (GLM, p <0.0001).
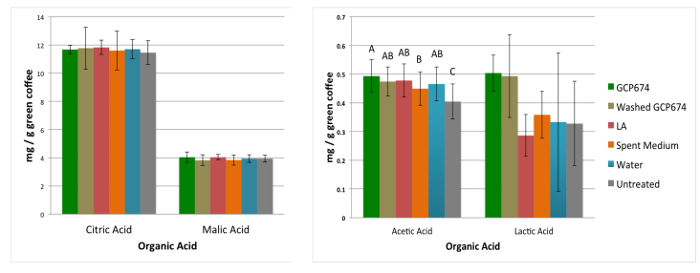
Figure 3: Impact of treatment on organic acid levels in green coffee. Treatment had no impact on (a) citric or malic acid levels in green coffee. However, all treatments significantly increased (b) acetic acid levels compared to no treatment controls (GLM, p <0.001). Increases in acetic acid levels were greatest with the microbe + medium treatment. Letters indicate significant difference by Tukey pairwise comparisons at 95%. Please click here to view a larger version of this figure.
The greatest increase was observed in the microbe + media treatment, which increased 0.25-fold (0.5 vs 0.4 mg/g coffee in GCP674 vs. NT). A similar trend was observed in lactic acid levels, though they were not significantly different. There were no significant changes or trends in the other acids tracked (citric and malic acid).
