Microglia isolated using CD11b Positive Selection kit II have high purity
Primary mouse microglia were isolated using the above mentioned protocol and plated on poly-D-lysine-coated coverslips to check the purity of isolation. Ten thousand cells were plated per well and immunocytochemical analysis was performed using ionized calcium-binding adaptor molecule 1 (Iba1) as a marker of microglia and glial fibrillary acidic protein (GFAP) as a marker of astrocytes to check for the purity of the isolated microglia. The isolated culture only expressed Iba1 without GFAP expression (Figure 2A), suggesting that the culture isolated was pure microglia. Unlike other previously published methods for primary microglial separation, which achieved ~97% purity, using this method we obtained ~>99% purity in half the time. To further validate the purity of this culture, we ran a Western blot20 for Iba1 and GFAP. Immunoblot analysis further revealed that this isolation from the mixed glial cultures was a nearly pure microglial culture (Figure 2B).
The modified isolation procedure does not have any auto-fluorescence from magnetic beads
The red channel cannot be used for immunocytochemistry (ICC) when using the previous CD11b isolation kit for microglial separation as mentioned by Gordon et al.15 because of the use of PE labeling during separation. Using this new PE-free isolation kit, we can use the red channel for immunocytochemical analysis (Figure 3). From this ICC, it is evident that this new method enables us to use all the channels for flow cytometry or other fluorescent imaging studies.
Isolated microglial cultures functionally respond to LPS stimulus
To verify that the isolated microglia are functionally active, we treated the cells with LPS, a widely used stimulant9 to activate microglia, for 24 h before probing for various pro-inflammatory factors. Classical microglial activation is accompanied by the release of nitrite and various pro-inflammatory cytokines into the media. Hence, we used multiple complementary assays to confirm the functional activity of our isolated microglia. First, we used the Griess assay to show that LPS dramatically induced nitrite secretion from the isolated microglia (Figure 4A). To further validate the activity of isolated microglia, we used qRT-PCR to show (messenger ribonucleic acid) mRNA levels of NOS2, another hallmark of microglial inflammation, significantly enhanced with LPS treatment (Figure 4B). Next, we used a bead-based multiplex assay, Luminex21, to verify that LPS significantly stimulated the secretion of pro-inflammatory cytokines from the microglial culture (Figure 5A). Furthermore, we verified via qRT-PCR analysis that LPS treatment enhanced the gene expression levels of several pro-inflammatory cytokines including IL-1β and TNFα (Figure 5B). All these data together suggest that primary microglia isolated using this newly refined method are functionally active and show a similar activation profile as primary microglia isolated using previously published methods.
Isolated microglia can be used for signaling studies
Using the previous methods of microglial isolation, running Western blot for signaling studies was difficult and infeasible due to low yield and purity. Previously, we have shown that Fyn, a Src family kinase, is involved in a pro-inflammatory signaling cascade in microglial cells9. Here we plated one million microglial cells in a 12-well plate and after 48 h, we collected the cells to run Western blots for native Fyn and Src kinases phosphorylated at tyrosine residue 416 (p-Src-Y416). We were able to detect both Fyn and p-Src-Y416 levels in our isolated microglial cells (Figure 6), showing that this method is applicable to Western blotting techniques for signaling studies.
The negative fraction from the microglial separation contains astrocytes that can be used for signaling studies
Astrocytes are key players in neuroinflammation, and understanding the signaling mechanisms behind astrocytic inflammation and neuron-astrocyte cross-talk is important19. Here, we show that the negative fraction from the microglial separation contains GFAP-positive astrocytic cells (Figure 7A). Also, our Western blot analysis for Fyn and p-Src-Y416 (Figure 7B) showed that both of these proteins can be detected in the negative fraction as well. These results together show that the negative fraction is an ideal preparation for astrocytic studies aimed at identifying proteins important for inflammatory signaling cascades.
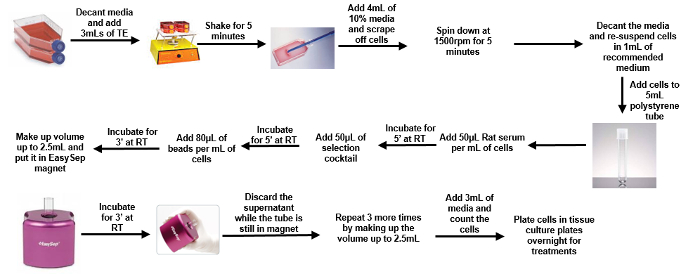
Figure 1: Schematic of Isolation of Microglial Cells from 1-2 Days-post-natal Pups. Please click here to view a larger version of this figure.
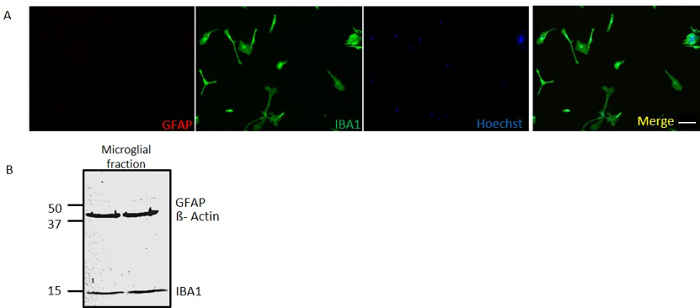
Figure 2: Microglia Isolated using CD11b-positive Selection Kit II Have High Purity. (A) Immunocytochemistry of isolated microglial culture probing for GFAP in red channel and IBA1 in green channel. (B) Immunoblot of isolated microglial culture probing for GFAP (~51 kDa), IBA1 (~15 kDa) and β-Actin (~42 kDa). Scale bar = 20 µm. Please click here to view a larger version of this figure.
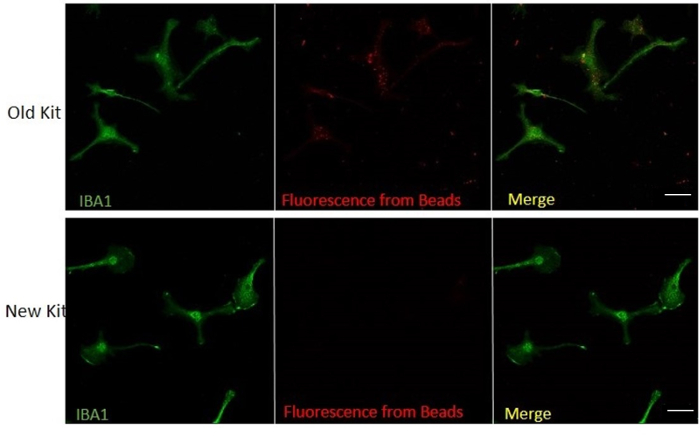
Figure 3: The Modified Isolation Procedure does not Have any Auto-fluorescence from Magnetic Beads. Immunocytochemistry of isolated microglial culture using the new isolation method and old (original) method probing for IBA1 in green channel and PE fluorescence in red channel. Scale bar = 30 µm. Please click here to view a larger version of this figure.
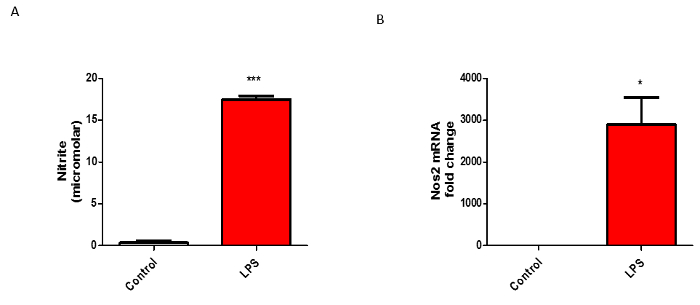
Figure 4: LPS Stimulation Increased Nitrite Production in Microglial Culture. (A) Isolated microglial cells treated with 1 µg/mL LPS for 24 h and the treatment medium were collected to determine nitrite release by Griess assay. (B) q-RT-PCR for Nos2 from isolated microglia treated with LPS for 24 h. The figures represent the mean ±SE from 2 or more independent experiments. Data were analyzed using Student's t-test (*p <0.05, **p <0.01, ***p <0.001). Please click here to view a larger version of this figure.
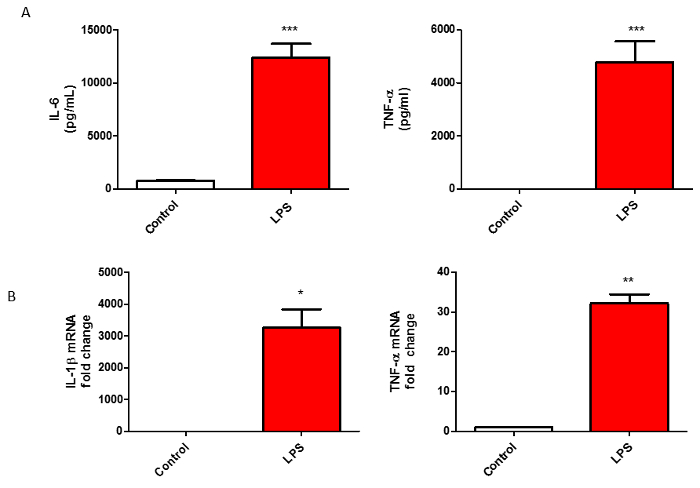
Figure 5: LPS-induced Pro-inflammatory Cytokine Release from Microglial Cells. (A) Luminex multiplex assay was performed on treatment medium collected from isolated microglial cells treated with 1 µg/mL LPS for 24 h. (B) q-RT-PCR for IL-1βand TNFα from isolated microglia treated with LPS for 24 h. The figures represent the mean ±SE from 2 or more independent experiments. Data were analyzed using Student's t-test (*p <0.05, **p <0.01, ***p <0.001). Please click here to view a larger version of this figure.

Figure 6: Isolated Microglia can be used for Signaling Studies. Western blot analysis shows that Fyn and p-Src-Y416 can be detected from microglia isolated with our newly refined method.
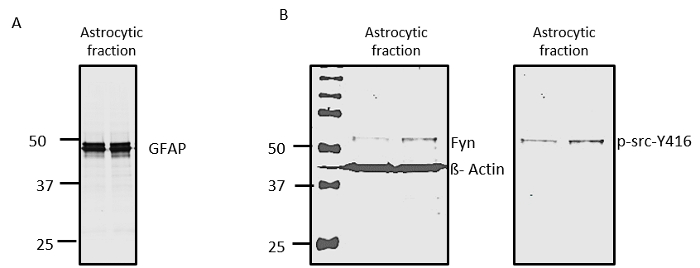
Figure 7: The Negative Fraction from the Microglial Separation Contains Astrocytes that can be used for Signaling Studies. (A) Western blotting shows that the negative fraction contains GFAP-positive cells. (B) Western blot analysis shows that Fyn and p-Src-Y416 can be detected from the GFAP-positive cells. Please click here to view a larger version of this figure.
| Parameters | Current method | Modified method | |
| Cost | $620 | $620 | |
| Time | 55 min | 25 min | |
| Fluorescence | Yes (red channel) | No | |
| Purity | ~97% | ~99% | |
Table 1: Comparative Analysis between the Refined Microglial Isolation Method and the Original Method of Isolation.
