To quickly create an inducible expression model, we made use of HEK293 cells that express Tetracycline Repressor protein (TR) (e.g., T-REx-293) and vectors that contain tetracycline operator sequences (TetO) between the CMV promoter and the multiple cloning site (e.g., pcDNA4/TO). When transfected into TREx-293 cells, the expression of the gene of interest in pcDNA4/TO is repressed, as TR binds to TetO. Adding doxycycline to the medium prevents TR-TetO interaction, thus allowing the expression of the transfected protein. To control the expression of rat TRPV1 (rTRPV1) we first inserted this channel's gene into a pcDNA4/TO vector using the above-described protocol. Next, T-REx-293 cells were transfected with the rTRPV1- pcDNA4/TO plasmid according to the steps in point 3 of the protocol. Following transfection, rTRPV1 expression was induced by incubating the cells in the presence of doxycycline (1 μg/mL) for 1 – 4 h. Importantly, different doxycycline concentrations can be used to achieve desired expression rate. We used electrophysiological and calcium imaging methods to evaluate rTRPV1 expression level at each time point by applying its agonist, capsaicin. As shown in Figure 1, using live-cell calcium imaging, intracellular calcium level after capsaicin application is increased gradually with longer induction times. Activation of uninduced cells may be attributable to a basal leak in the TR activity, overexpression of the plasmid, or residual tetracycline in the cells media components. Next, we recorded currents from cells using the whole-cell configuration of the patch clamp technique applying voltage ramps between −80 mV to +80 mV. Figure 2 shows that higher current amplitudes are obtained with longer induction times. The saturation in current amplitude at 4 h induction is consistent with the saturation seen in calcium response (Figure 1). Finally, we analyzed rTRPV1 expression levels in the outside-out configuration of the patch-clamp technique. One of the major difficulties in studying ion channel structure-function is reaching expression levels suitable for single-channel analysis. Based on the published unitary rTRPV1 conductance4, the recorded current amplitude was used to determine the number of channels in the excised patch. As shown in Figure 3, the number of channels in the patch increases proportionately to the doxycycline incubation time. After one hour of induction, we were not able to detect any channel activity (0 out of 8). However, in both two- and three-hour time points we recorded single channel activity in similar success rates. Of note, while in two hours most patches did not respond, in three hours most patches showed single-to-multi -channel activity. The chances of recording multiple channels after three hours of induction is high, thus the optimal induction time to record current from a single channel is between two to three hours under the presented conditions. Together these results demonstrate that expression of ion channels can be tightly controlled and regulated after transient transfection using this protocol.
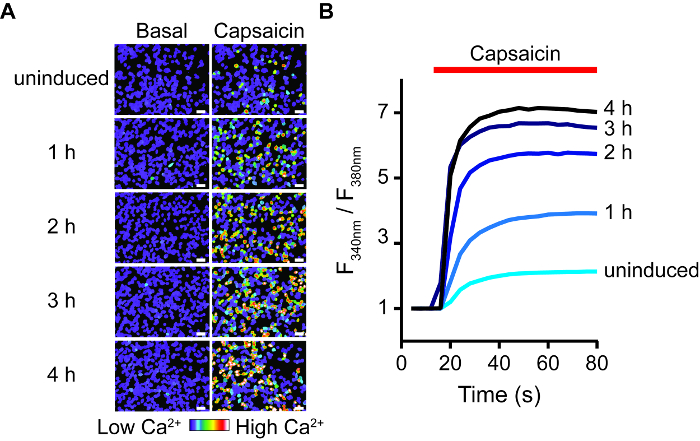
Figure 1. Response of TRPV1 Activation Increases according to Induction Time, as Visualized through Calcium Imaging. (A) Pseudo-colored images of T-REx-293 cells transiently expressing rTRPV1, before ('Basal') and after capsaicin (2 μM) application. White bars represent 30 μm. Scale bar indicates levels of intracellular calcium. (B) Changes with time of intracellular calcium levels in transfected T-REx-293 cells treated as shown in A. Each graph represents an average of 50 capsaicin sensitive cells. Note the stepwise increases in capsaicin responses in relation to induction time. Please click here to view a larger version of this figure.
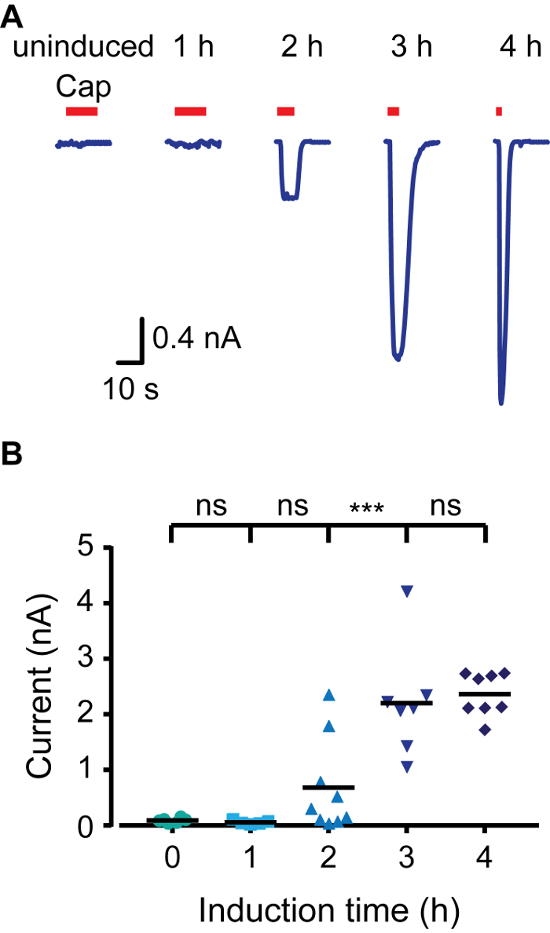
Figure 2. TRPV1 Current Increases as a Result of Increasing Induction Time in Whole Cell Patch Clamp Recordings. (A) Whole-cell recordings from T-REx-293 transiently transfected with rTRPV1 at a holding potential of -40 mV. Cells were induced with doxycycline (1 μg/mL) for the indicated time and then exposed to capsaicin ('Cap'; 1 μM; red bar). Shown is a representative trace of 6 – 11 independent recordings. (B) Mean/scatter-dot plot representing the whole-cell amplitude evoked as shown in A. Statistical significance between groups was determined with ANOVA with multiple comparisons, where *** represents p ≤0.001 and ns- not statistically significant (n = 6 – 11 cells). Please click here to view a larger version of this figure.
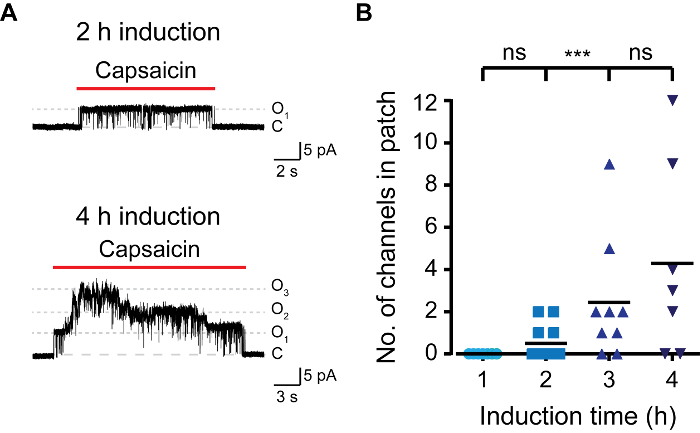
Figure 3. Success Rate of Single Channel Patch in TRPV1 Recordings is Dependent on Induction Time. (A) Outside-out recordings from T-REx-293 transiently transfected with rTRPV1 at a holding potential of +50 mV. Cells were induced with doxycycline (1 μg/mL) for the indicated time and were exposed to capsaicin ('Cap'; 1 μM; red bar). Shown is a representative trace of 6 – 9 independent recordings. (B) Mean/scatter-dot plot representing the number of channels in the excised patch as shown in A. Statistical significance between groups was determined with ANOVA with multiple comparisons, where *** represents p ≤0.001 and ns- not statistically significant (n = 6 – 9 cells). Please click here to view a larger version of this figure.
