Figure 1 shows a schematic representation of the experimental arrangement of the spectrometer, glovebox, ATR accessory, potentiostat and gas flow system used for PFIRE measurements. Figure 2 shows a representative drawing of the spectroelectrochemical cell.
Figure 3 shows absorbance spectra of drop-cast Hyd1-modified particles, with the experimental buffer (a mixed buffer system, described in 3.7, pH 6.0) flowing through the spectroelectrochemical cell. The surface coverage of Hyd1 is particularly high in the example shown in Figure 3, with an amide II band intensity of ~235 mO.D. and minimal 'bulk' water as evidenced by the magnitude of the O-H stretching region (~3,000 – 3,600 cm-1) relative to the band at ~1,640 cm-1, which is a convolution of the amide I band of Hyd1 and the water H-O-H bend. Additional bands due to the protein can be seen in the C-H stretching region (ca 2900 cm-1). The broad band centered around 2,100 cm-1 is a combination band of the H-O-H bending vibration with a set of lower energy libration bands, which are restricted rotations of H2O molecules due to the hydrogen bonding network in liquid water. The νCO band of the oxidized, inactive, Ni-B state of the active site is clearly evident at 1,943 cm-1, even without baseline correction, and νCN features are clearly visible between 2,050-2,100 cm-1. At high Hyd1 coverages, much of the microporous structure of the carbon black film57 becomes blocked by enzyme and therefore the 'bulk' water concentration is lowered during PFIRE measurements.
The spectra in Figure 3 show that, before activation, Hyd1 films contain Hyd1 in oxidized, inactive states. Activation overnight at −0.8 V vs SCE under a H2 atmosphere leads to formation of reduced, catalytically active states as demonstrated in Figure 4 which shows an activated (reduced) minus as-prepared (oxidized) difference spectrum of Hyd1. Difference spectra of Hyd1 can be most clearly interpreted using the νCO region. Each unique state of the active site has only one CO band compared to two CN bands and therefore the νCN region is intrinsically more complicated, with many overlapping bands. The difference spectrum in Figure 4 shows that activation leads to loss (negative absorption bands) of oxidized, inactive Ni-B and a small amount of Ni-SI (the most oxidized 'active' state) that was present in the as-prepared Hyd1 film. These are replaced by 'active' states of Hyd1; Ni-C, Ni-R and Ni-L. Note that there are two forms of both the Ni-R and Ni-L states, as evidenced by the two νCO bands observed for these species in Figure 4. The observation of multiple Ni-R and Ni-L states is in agreement with other NiFe hydrogenases.48,58,59
A key verification of the PFIRE method is that the cyclic voltammograms recorded of Hyd1 inside the spectroelectrochemical flow cell show similar catalytic waveshapes to those recorded on a planar rotating disc electrode.55 In practice this means that the mass transport of substrate (H2) and product (H+) to/from immobilized Hyd1 in the spectroelectrochemical cell is efficient at the flow rates used during PFIRE measurements. The effect of flow rate on the catalytic waveshape is shown in Figure 5, which shows successive voltammograms recorded under a H2 atmosphere (1 bar) as the solution flow rate through the spectroelectrochemical cell is increased. In all cases the overpotential for H2 oxidation by Hyd1 is identical (red shaded rectangle), but the extent of oxidative inactivation (hysteresis between the current during the oxidative and reductive sweeps at potentials above ca 0 V vs SCE) and the maximum H2 oxidation current are dependent upon the solution flow rate. At flow rates above 52 mL/min (light grey voltammogram) the catalytic waveshape is insensitive to further flow rate increases.
Figure 6 compares the relative intensities of the νCO band of the Ni-B state after initial activation and anaerobic re-oxidation at 0 V vs SCE (under a Ar atmosphere, as described in 4.3) and after anaerobic oxidative inactivation under an Ar atmosphere at 0 V vs SCE after 48 hr of continuous experiments (as described in 5.1). No loss of active site band intensity is observed during the measurement, and all the Hyd1 sample responds to the applied potentials.
Figure 7 demonstrates the baseline correction procedure used throughout this work. The absolute spectrum of the Hyd1 sample in the active site region (Figure 7a) contains significant curvature due to water. In fact, the requirement to use water as a solvent is a problem for most applications of IR spectroscopy in the life sciences. The second derivative of the absolute spectrum (Figure 7b), calculated using Origin software with Savitsky-Golay smoothing over a 9 point window, can be used to identify sharp bands of the Hyd1 active site against the curved background. The identification of approximate peak positions using a second derivative spectrum allows baseline anchor points to be placed in regions of the absolute spectrum that are free of active site bands (circles in Figure 7a). A cubic spline function is then fitted through these anchor points to create a baseline function that can then be subtracted from the absolute spectrum to give a baseline-corrected spectrum containing only peaks arising from the Hyd1 active site (Figure 7c).
Figure 8 shows the results of a PFIRE measurement on Hyd1, under both non-turnover (Ar atmosphere) and turnover (H2 atmosphere) conditions at a range of potentials.36 The current-time traces (Figure 8a) report on the catalytic current at each applied potential and remain at close to zero current under non-turnover conditions (Ar atmosphere). The partial spectroelectrochemical redox titration in Figure 8b therefore reports on the equilibrium redox behavior of the active site, showing the distribution of states expected at each potential in the absence of catalytic turnover. The spectra in Figure 8c were recorded under turnover conditions (under a H2 atmosphere) and therefore represent the steady-state distribution of active site states present during catalytic H2 oxidation by Hyd1. That steady-state conditions have been achieved is confirmed by the fact that the catalytic H2 oxidation current (Figure 8a, H2) remains constant as a function of time at −0.199 V and −0.074 V vs SHE; the monotonic decay in current at +0.356 V vs SHE is due to the well-known anaerobic oxidative inactivation of Hyd1.55 The distribution of active site states is clearly different under Ar and H2 at all potentials where Hyd1 performs catalysis (Figure 8b, spectra at −0.199, −0.074 and +0.356 V vs SHE). The spectra recorded under Ar and H2 are virtually identical at −0.594 V vs SHE, however, and this represents an important test of experimental consistency; Hyd1 does not reduce H+ at a significant rate at pH 6.0 (the current in Figure 8a under both Ar and H2 is close to zero), and the spectra at −0.594 V are therefore expected to be the same.
Figure 9 demonstrates the anaerobic oxidative inactivation of Hyd1 via the formation of Ni-B from Ni-SI during H2 oxidation at +0.356 V.36 Spectra were recorded during the grey time intervals noted on the current-time trace in Figure 9a. At −0.074 V Hyd1 does not undergo oxidative inactivation, and the distribution of active site states remains constant throughout the entire potential step. This is demonstrated by the spectra in Figure 9bi, which reports the absolute baseline-corrected spectrum at the beginning of the −0.074 potential step, and Figure 9bii which was recorded at a later time during the potential step and is reported as a difference spectrum relative to Figure 9bi. The spectra in Figure 9biii and biv are also reported as difference spectra relative to Figure 9bi, and show gradual conversion of Ni-SI to Ni-B during high potential inactivation, consistent with the monotonic decrease in current shown at +0.356 V in Figure 9a.
Spectra recorded at a range of solution pH give insight into the proton transfer steps during the Hyd1 catalytic cycle.43 Figure 10a shows PFIRE spectra recorded on the same Hyd1 film at pH 3.0 and pH 9.0, using solution flow in the spectroelectrochemical cell to exchange the experimental buffer. The relative concentrations of the Ni-C and Ni-L states are clearly different in these two spectra. By varying the applied potential under non-turnover conditions the potential dependence of the NiC and Ni-L states can be determined rapidly at a range of pH values (Figure 10b), and both states are found to be isopotential over a wide pH range. (Note that the peak absorbances in Figure 10b show only the Ni-C and Ni-L states for clarity, full redox titrations of Hyd1 have been reported by Hidalgo et al.)36 A pH titration of the concentrations of Ni-C and Ni-L can then be extracted, taking the peak absorbance at potentials where the total Ni-C and Ni-L concentration is at its maximum at each pH (Figure 10c). In this way, and in conjunction with EPR data, a pH equilibrium between the Ni-C and Ni-L states was identified.43
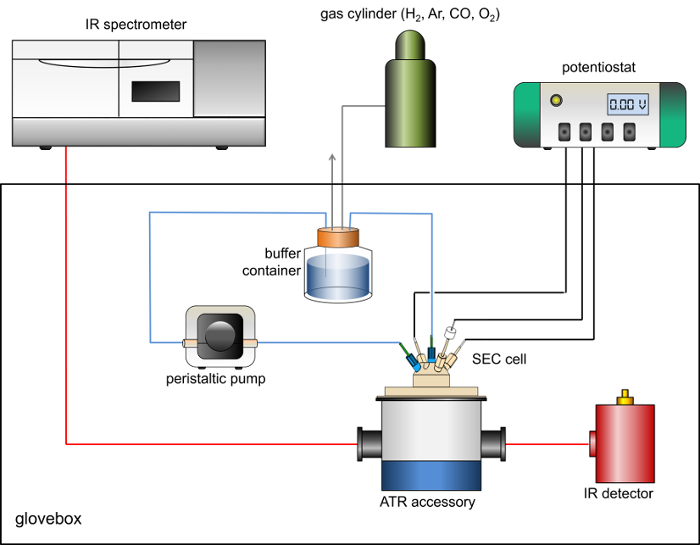
Figure 1: Schematic of the arrangement of IR spectrometer, anaerobic glovebox, ATR accessory, MCT detector, gas flow system and potentiostat used for PFIRE measurements. Please click here to view a larger version of this figure.
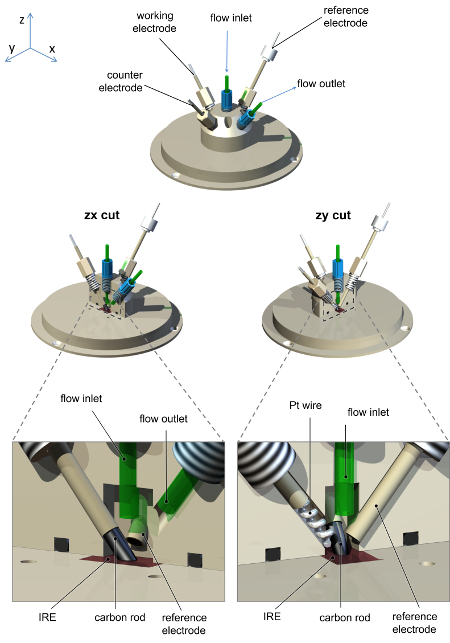
Figure 2: Schematic diagram of the spectroelectrochemical cell used for PFIRE measurements, showing the arrangement of electrodes and solution inlet/outlet connections. The cell and baseplate are machined from polyether ether ketone (PEEK), with screw holes for a carbon rod working electrode connection, Pt wire counter electrode, saturated calomel reference electrode, and solution inlet and outlet. The saturated calomel reference electrode construction is as previously reported.36 Please click here to view a larger version of this figure.
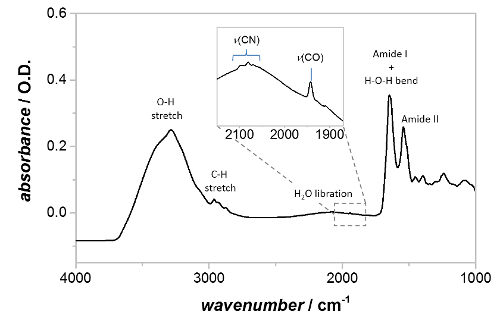
Figure 3: Absorbance spectrum of Hyd1-modified carbon black particles, deposited onto the IRE and rehydrated with buffer. The positions of the amide I band, amide II band, and Hyd1 active site region are shown, along with additional features due to C-H stretching vibrations and solvent water. The inset shows a magnified view of the active site region, with the vCO and vCN bands labeled. ‘As-prepared’ particles contain Hyd1 mainly in the oxidized, inactive Ni-B state. Please click here to view a larger version of this figure.
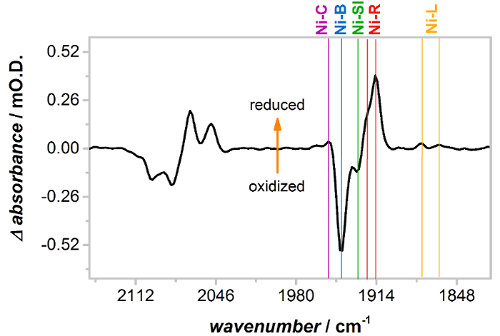
Figure 4: Activation of Hyd1 at −0.8 V vs SCE under a H2 atmosphere, presented as a reduced minus oxidized difference spectrum. Upon low potential activation oxidized, inactive Ni-B (and a small concentration of Ni-SI) converts to more reduced, active states Ni-C, Ni-R and Ni-L. Note that Hyd1 has two distinct sub-states of Ni-L and Ni-R. Please click here to view a larger version of this figure.
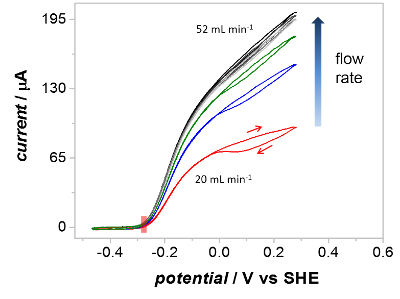
Figure 5: The effect of solution flow rate on the waveshape of catalytic cyclic voltammograms recorded in the spectroelectrochemical cell. Voltammograms were recorded at increasing flow rates of H2-saturated buffer as indicated. At a flow rate of 20 mL/min (red) the voltammogram shows significant inactivation above 0 V vs SHE on the forward scan. At flow rates above 52 mL/min the extent of inactivation is vastly lower and the current is independent of flow rate at all potentials. Other parameters: 1 bar H2, 10 mV/s scan rate. Please click here to view a larger version of this figure.
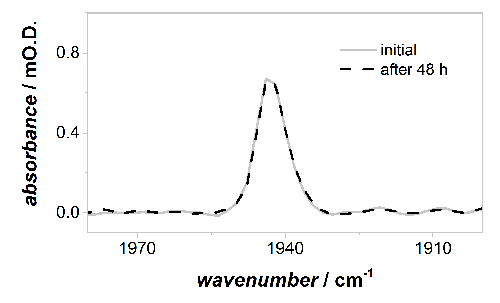
Figure 6: Baseline corrected IR spectra in the active site νCO region of oxidized, inactive Ni-B state at 0 V vs SCE. There is no measurable loss of active site intensity during 48 h of continuous PFIRE measurements, and therefore Hyd1 is adsorbed robustly on the carbon black particles. Please click here to view a larger version of this figure.
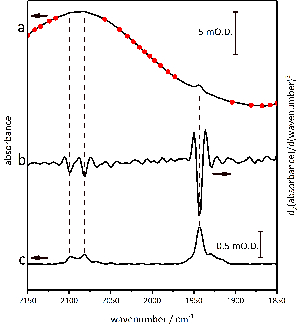
Figure 7: Details of the baseline correction procedures used for data handling. Baseline anchor points are placed onto the absolute absorbance spectrum in the active site region (a), taking care to avoid any νCO and νCN peaks identified from a second derivative analysis (b). The resulting baseline corrected spectrum is shown in (c). Please click here to view a larger version of this figure.
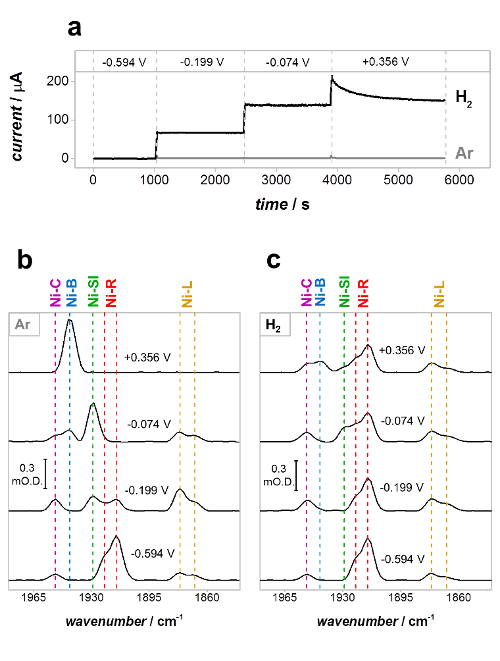
Figure 8: PFIRE measurements on Hyd1 under non-turnover (Ar) and turnover (H2) conditions. (a) Current-time traces of Hyd1 in the spectroelectrochemical cell in Ar-saturated (gray) and H2-saturated (black) buffer; (b), (c) PFIRE spectra showing the νCO region at each potential under Ar (b) and H2 (c). Potentials quoted in V vs SHE. Reproduced with permission from Hidalgo et al.35 Please click here to view a larger version of this figure.
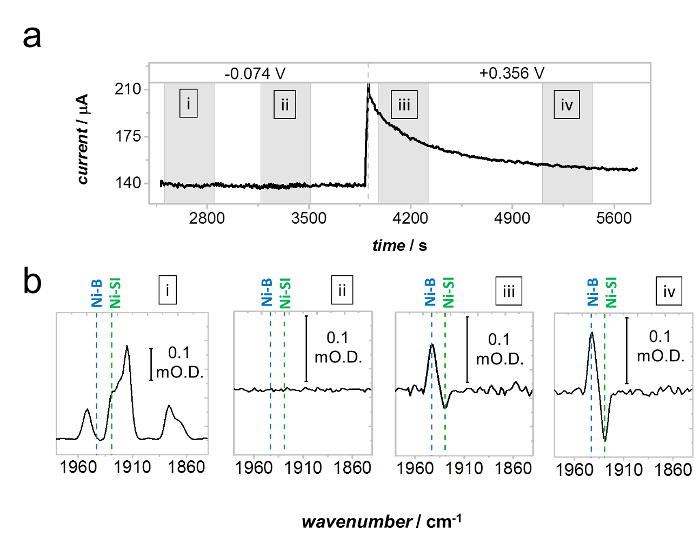
Figure 9: Anaerobic inactivation of Hyd1 via formation of Ni-B from Ni-SI. (a) Current-time trace under a H2 atmosphere, showing a stable electrocatalytic current at −0.074 V and slow anaerobic inactivation (monotonic decrease in current) at +0.356 V vs SHE. (b) Spectra recorded during the gray shaded regions marked in (a). Spectrum bi is a baseline corrected spectrum recorded at the beginning of the −0.074 V potential step. Spectrum bii, recorded at a later time during the −0.074 V step, is reported as a difference spectrum relative to bi and shows that no change in distribution of active site states occurs, consistent with the stability of the potential at −0.074 V. Spectra biii and biv are also reported as difference spectra relative to bi and show gradual conversion of Ni-SI to Ni-B during anaerobic inactivation at +0.356 V vs SHE. Reproduced with permission from Hidalgo et al. 35 Please click here to view a larger version of this figure.
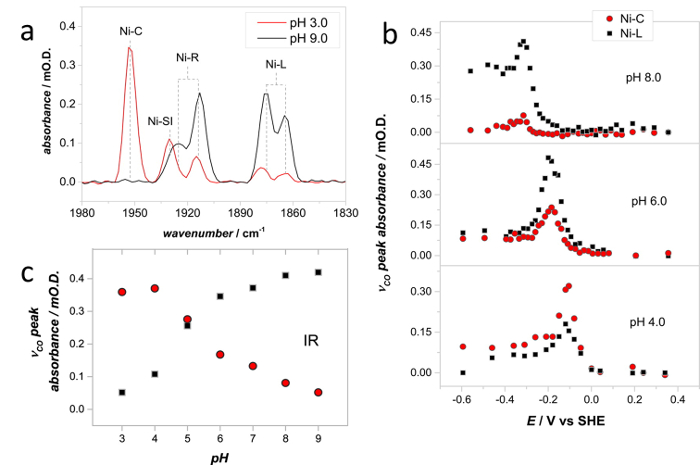
Figure 10: Spectra recorded at a range of solution pH give insight into the proton transfer steps during the Hyd1 catalytic cycle. (a) IR spectra showing the νCO region of Hyd1, recorded at pH 3.0 (-54 mV vs SHE) and pH 9.0 (-334 mV vs SHE). (b) Spectroelectrochemical titrations were carried out to determine the potential at which the Ni-C and Ni-L concentrations are at a maximum at a range of solution pH values. Only the Ni-C and Ni-L concentrations are shown for clarity, for a full spectroelectrochemical titration of Hyd1 see Hidalgo et al.36 (c) pH dependence of the relative concentration of Ni-C and Ni-L, as determined from a series of experiments such as those shown in (b). Spectra were recorded at 20 °C. Adapted with permission from Murphy et al.42 Please click here to view a larger version of this figure.
