In this in vitro phagocytosis assay with long-term live-imaging, we used synaptosomes from adult mouse brain homogenates, which were separated in the gradient solution between 23% gradient solution and 10% gradient solution by ultracentrifugation (Figure 3). After the preparation, synaptosomes exposed PS in their outer membrane (Figure 4), suggesting that they lost their function and could be recognized by PS receptors in astrocytes and microglia. As shown in Figure 5, pH indicator-conjugated synaptosomes emitted bright red fluorescence when they were engulfed by astrocytes. Real-time comparison of the engulfment and degradation capacities of glial cells is possible by taking multiple images of a ROI every 1 or 2 h (Supplementary Movie 1, Supplementary Movie 2). With this method, we demonstrated different kinetics of astrocyte- and microglia-mediated phagocytosis (Figure 6). To quantify phagocytosis of glial cells, the area (µm2) of red fluorescence signal was measured, which is referred to phagocytic index in Figure 6B and Figure 7. Although astrocytes appeared to be efficient in phagocytosing large amounts of pH indicator-conjugated synaptosomes, microglia were faster at engulfing and degrading synaptosomes (Figure 6B). Microglia showed maximum pH indicator intensity at 26 h after pH indicator-conjugated synaptosome treatment, whereas astrocytes showed their maximum at 45 h (p-value < 0.05, two-way ANOVA between astrocyte with 1X ACM and microglia with 1X ACM). Likewise, microglial cells showed a 20.7% reduction in total pH indicator intensity 40 h after the peak point, whereas astrocytes showed a 17% reduction in total pH indicator intensity during the same time period. Interestingly, our data showed that astrocyte-secreted factors, which were contained in ACM, are essential for increasing both astrocyte- and microglia-mediated phagocytosis (Figure 6B). Astrocytes have been shown to release bridging molecules, such as MFGE8, GAS6, and ProteinS, which can bridge and induce interactions between phagocytic receptors and "eat-me" signals such as PS23. As mentioned above, astrocytes eliminate synapses via the MERTK and MEGF10 pathways4. MEGF10 is only expressed by astrocytes in the brain and participates in synapse engulfment through recognizing "eat-me" signals with unknown identities. In agreement with previous findings, the assay showed that compared with wild-type (WT) mouse astrocytes, Megf10 knock-out (KO) mouse astrocytes possessed significantly impaired phagocytic capacity (Figure 7). Compared with WT astrocytes, Megf10 KO astrocytes showed an approximately 40% reduction in total pH indicator intensity at the peak point (31 h) (Figure 7).
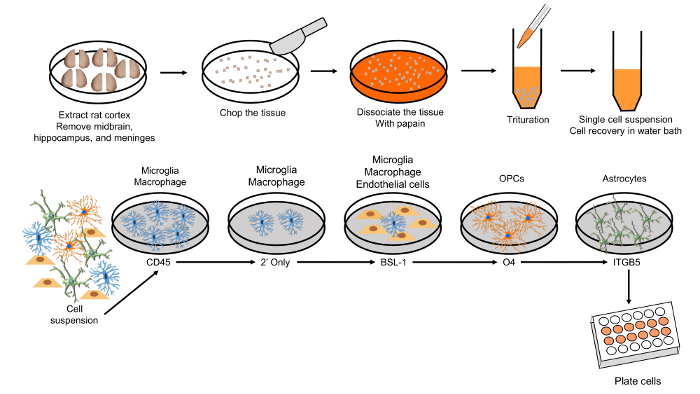
Figure 1. Schematic of astrocyte purification using immunopanning methods. Please click here to view a larger version of this figure.
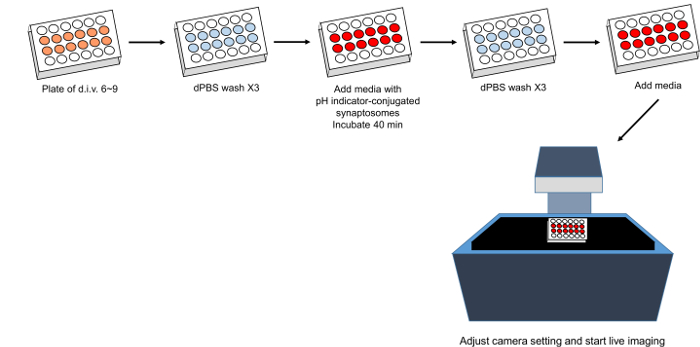
Figure 2. Schematic of phagocytosis live-imaging assay. Please click here to view a larger version of this figure.
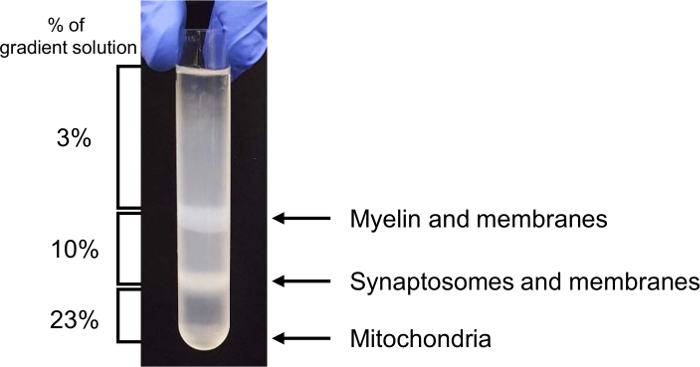
Figure 3. Representative brain homogenate fractionation in the gradient solutions. Please click here to view a larger version of this figure.
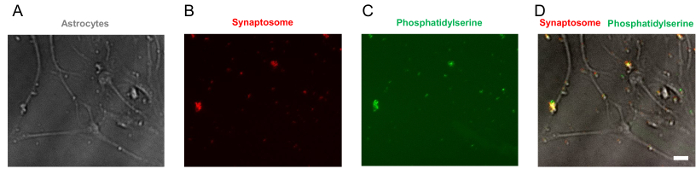
Figure 4. Representative images of PS-exposed synaptosomes. (A) A bright field image of astrocytes with synaptosomes. Synaptosomes are attached to astrocytes. (B) A fluorescent image of tdTomato-positive synaptosomes, which are purified from tdTomato-expressing mouse brains. (C) pSIVA binds to PS, which is exposed to outer membrane of synaptosome, and emits green fluorescence. (D) PS detected by pSIVA (green) is co-localized with tdTomato-positive synaptosomes (red). Scale bar: 20 µm Please click here to view a larger version of this figure.
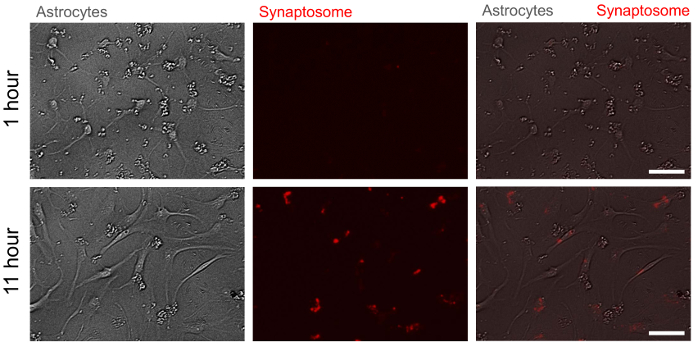
Figure 5. Representative bright field and fluorescent images of astrocytes with pH indicator-conjugated synaptosomes at two time points. At 11 h after treatment (bottom panel), pH indicator-conjugated synaptosomes are engulfed by astrocytes and emit red fluorescence whereas they do not at 1 h after treatment (upper panel). Scale bar: 50 µm. Please click here to view a larger version of this figure.
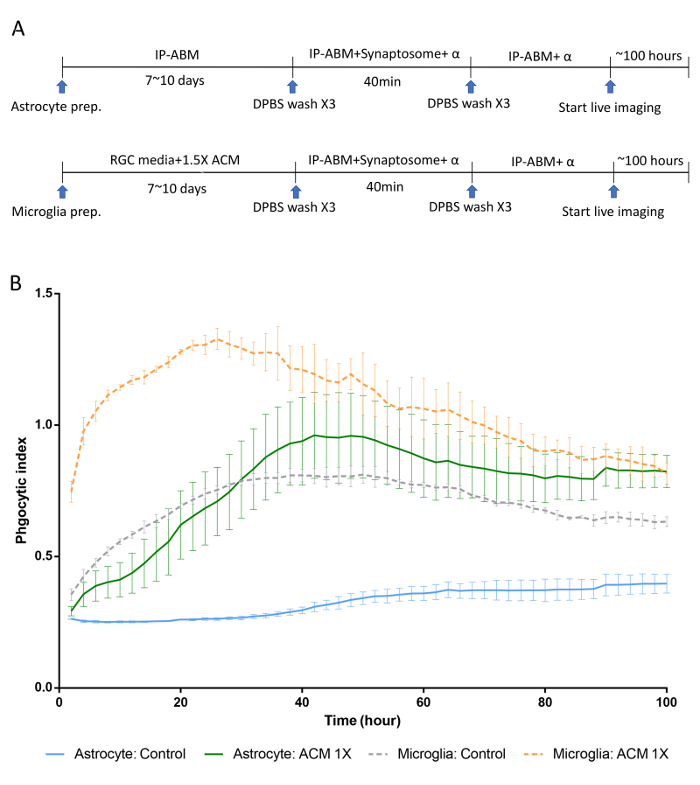
Figure 6. Phagocytic kinetics of rat astrocytes and microglia through long-term live-imaging. (A) A schematic diagram of an in vitro phagocytosis assay using purified astrocytes and microglia along with pH indicator-conjugated synaptosomes. Note that before live images are taken, unbound synaptosomes are washed away after 40 min of incubation. (B) Representative graphs showing the engulfment and degradation kinetics of astrocytes and microglia. ACM, which contains astrocyte-secreted factors, significantly enhances both astrocyte- and microglia-mediated synaptosome uptake. Astrocyte: Control vs. Astrocyte with ACM 1X, ****, Tukey's multiple comparisons test. Microglia: Control vs. Microglia with ACM 1X, ****, Tukey's multiple comparison test. Error bars indicate S.E.M. *p ≤ 0.05, **** p ≤ 0.0001, two-way ANOVA. Please click here to view a larger version of this figure.
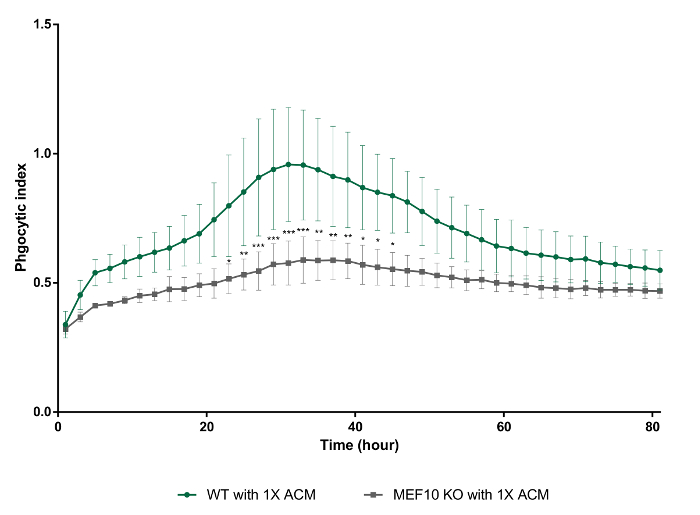
Figure 7. Decreased phagocytic capacity of Megf10 KO mouse astrocytes with 1X ACM compared with that of WT mouse astrocytes with 1X ACM. WT vs. Megf10 KO astrocytes with 1X ACM, ****, two-way ANOVA. Error bars indicate S.E.M. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001 two-way ANOVA. Please click here to view a larger version of this figure.
Table 1: Solution recipes. Please click here to download this file.
Supplemental Movie 1. A representative live-imaging video showing the phagocytosis of pH indicator-conjugated synaptosomes by astrocytes. Please click here to download this file.
Supplemental Movie 2. A representative live-imaging video showing phagocytosis of pH indicator-conjugated synaptosomes by microglial cells. Please click here to download this file.
