zFRAP analysis with eGFP-H2B
Properly produced mRNA encoding eGFP-H2B, which is seen as a shifted band caused by poly A tailing (Figure 2A), was injected into the cytoplasm of zygotes with a 2nd polar body at 1 – 3 h after insemination (Figure 2B). Eight to 12 h after insemination, zygotes with 2 pronuclei showing the expression of eGFP-H2B were collected and subjected to zFRAP analysis (Figure 2C). The FRAP analysis consists of bleaching and recovery steps. At the pre-bleaching phase, the signal of eGFP-H2B could be observed (Figure 2D-1), but once bleached, the signal declined to a negligible level (Figure 2D-2). If bleaching was successful, a dark hole as large as the ROI appeared soon after bleaching. After bleaching, the intensity of the eGFP-H2B fluorescence signal recovered slightly; the fluorescent signal gradually increased because of the inflow of the unbleached fraction of eGFP-H2B into the ROI from the unbleached area in the nucleoplasm (Figure 2D-3). For confirmation of the success of zFRAP analysis of chromatin looseness, it is recommended to use parental asymmetry of chromatin looseness; male pronuclei showed a faster recovery curve and more mobile fractions than those of females (Figure 2E and F). After zFRAP analysis, washed and cultured zygotes in CZB media, could develop to the blastocyst stage without significant damage (Figure 2G and H). Even if strongly bleached for a longer time, the zygotes could still develop into the blastocyst stage as well7.
Analysis of the full-term development of zFRAP-analyzed zygotes
To analyze the full-term development of embryos derived from zFRAP-analyzed zygotes, the embryos at the two-cell stage were transferred into the oviducts of pseudopregnant mothers. As controls, intact embryos, and one injected with mRNA but not zFRAP-analyzed were prepared. The birth rate of zFRAP-analyzed embryos (41.0%) was slightly but significantly lower than that of intact embryos (62.2%), but was the same as that of the no zFRAP control (52.6%) (Table 2). Thus, zFRAP-analysis seems to be slightly detrimental to full-term embryonic development. However, importantly, the pups derived from zFRAP-analyzed zygotes seemed healthy and fully developed (Figure 3).
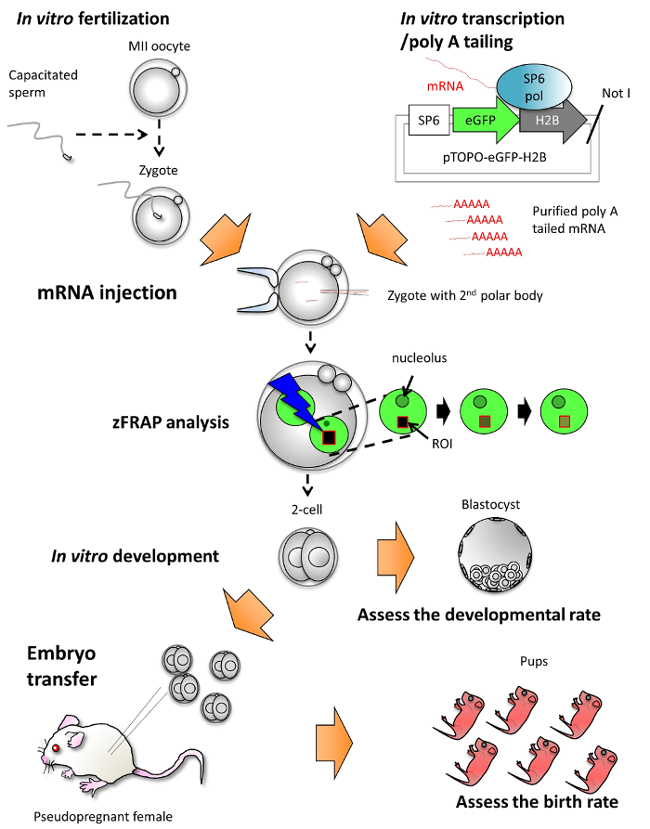
Figure 1: A schematic illustration of the flow of procedures for the experiments. In vitro fertilization: Freshly collected metaphase II (MII) oocytes were inseminated with capacitated sperm. In vitro transcription: Messenger RNA (mRNA) encoding eGFP-fused histone H2B (eGFP-H2B) were transcribed from the SP6 promoter of the linearized pTOPO-eGFP-H2B3 with Not I. The eGFP-H2B mRNA subjected poly A tailing and then purification. The purified eGFP-H2B mRNA is used in mRNA injection. mRNA injection: The prepared eGFP-H2B mRNA is injected into the cytoplasm of the zygotes with 2nd polar body. zFRAP analysis: The eGFP-H2B-expressing zygotes were subjected to zFRAP analysis. The region of interests (ROI; red rectangle) was bleached with a strong laser and then the eGFP-H2B signal declined to a negligible level. After bleaching, the intensity of the eGFP-H2B signal gradually increased. In vitro development: After zFRAP analysis, the zygotes could develop into the blastocyst stage. Embryo transfer: At two-cell stage, the zFRAP-analyzed embryos were transferred into the oviducts of pseudopregnant female mice. 18 days later, healthy live pups could be obtained from zFRAP-analyzed embryos. Please click here to view a larger version of this figure.
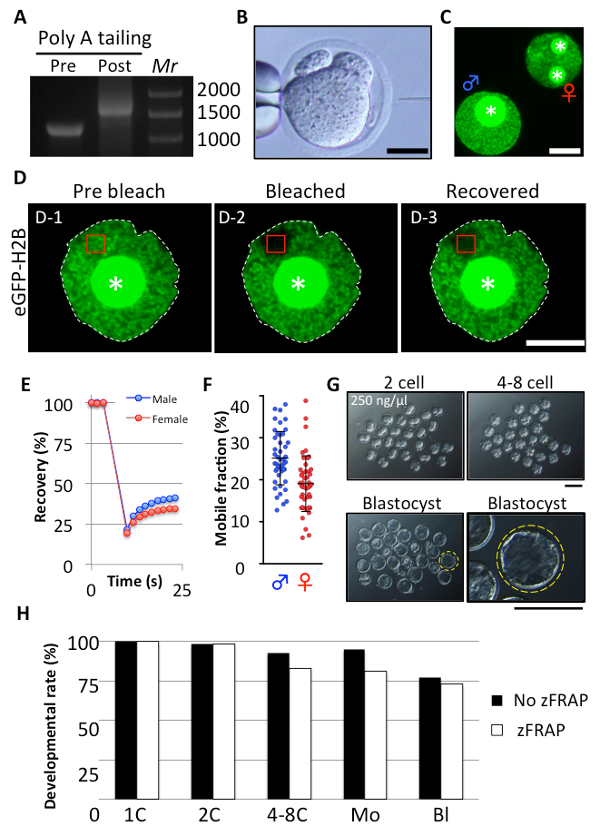
Figure 2: zFRAP analysis of zygotes with eGFP-H2B. (A) Representative image of properly prepared mRNA by in vitro transcription and poly A tailing. Lane 1: pre-poly A tailing; lane 2: post poly A tailing. (B) mRNA encoding eGFP-H2B was injected into the cytoplasm of zygotes with a 2nd polar body 1 – 3 h post insemination (hpi). Scale bar = 25 µm (C) eGFP-H2B-expressing zygotes were collected at 8 – 12 hpi. White asterisks show nucleolus precursor bodies (NPB). Scale bar = 10 µm (D) eGFP-H2B expression was detected in the entire nucleoplasm even in NPB at the pre-bleaching phase (D-1), soon after bleaching (D-2), and after recovery (D-3). The nuclear membranes (white dotted lines) and NPB (asterisks) are indicated. Scale bar = 10 µm. (E, F) Recovery curve and mobile fractions were obtained from 45 zygotes in 5 independent experiments. Blue and red indicate male and female, respectively. In recovery curves, single circles indicate the measuring point. In scatter plots, single dots indicate the score of mobile fractions obtained from pronuclei. (G) Representative images of the preimplantation development of zFRAP-analyzed zygotes are shown. Two-cell, 4-8 cell, and blastocyst stage embryos at 24, 48, and 96 hpi, respectively. The yellow circle indicates the well-developed blastocyst. This blastocyst is enlarged and shown on the right panel. Scale bar = 100 µm. (H) Bar graph of developmental rates of zFRAP-analyzed embryos. Bars indicate zFRAP-analyzed (zFRAP) embryos (white) and control embryos (black) injected with mRNA but no zFRAP analysis (No zFRAP). Data shown are from three independent experiments, examining at least 57 embryos in total. Two-cell, 4-8 cell, morula (Mo), and blastocyst (Bl) stages were observed at 24, 48, 72, and 96 hpi, respectively. This figure has been modified from [Ooga et al 2017]7. Please click here to view a larger version of this figure.
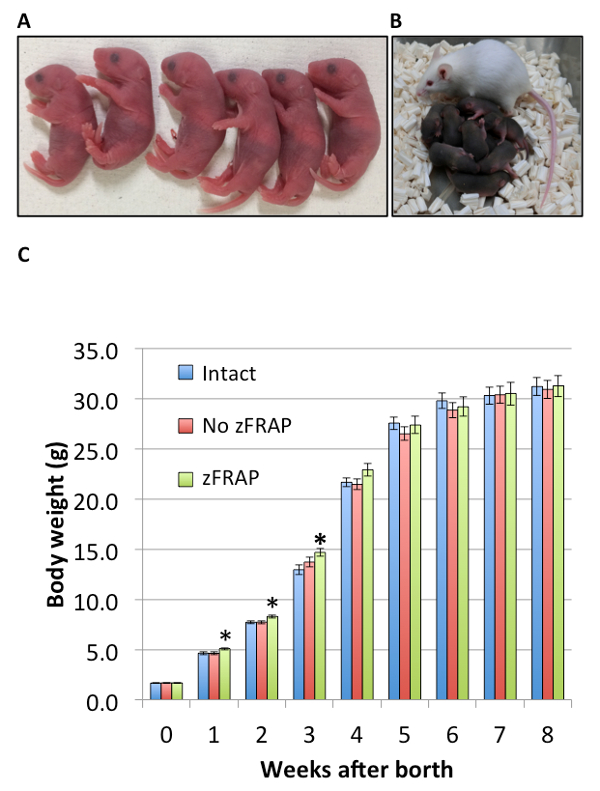
Figure 3: The healthy growth of pups derived from the FRAP-analyzed zygotes. (A) The pictures of pups derived from zFRAP-analyzed zygotes are shown. (B) Normal growth was observed during the nursing of the zFRAP-analyzed pups. (C) The graph indicates the weight of pups derived from intact embryos (blue), unbleached control embryos (red), and zFRAP-analyzed embryos (green). Body weights of 29 pups derived from intact embryos, 24 from no-zFRAP embryos, and 15 from zFRAP-analyzed embryos over an 8-week period. Values indicated by asterisks are significantly different from the intact control. This figure has been modified from [Ooga et al 2017]7. Please click here to view a larger version of this figure.
Supplemental Figure 1: Graphical user interface for FRAP analysis. (A) An overview of the graphical user interface was shown. (B) Enlarged images for each window were shown. (1) The Acquisition Setting window is used for setting the image acquisition condition. (2) The Stimulus Setting window is used for setting the bleaching condition. This window can be opened by clicking the button on the Image acquisition window. (3) The Image Acquisition Control window is used for the FRAP analysis. (4) The Live View shows the present image (the present image appears after clicking Focus x2 or XY button). The red, green, and light blue rectangles show the ROI (region of interest), REF (reference), and BG (background), respectively. (5) The 2D View shows the FRAP-analyzed image and appears after clicking the "Series Done" button. In this case, a FRAP-analyzed male pronucleus is shown as an example. Three rectangles with 8 dots indicate that these regions are selected. (6) The Live Plot shows the present fluorescence intensity at each region. The X and Y axes indicate time (ms) and fluorescence intensity, respectively. (7) The Series Analysis shows the tracks of fluorescence intensity at each region and appears after clicking Series Analysis button on the 2D View window. Please click here to download this file.
Supplemental Excel File 1: Example data and data calculation. (Sheet 1) An example data for a male pronucleus is shown. No. indicates the consecutive number of the image. The row intensities for each region (ROI, Ref, and BG) were indicated. (Sheet 2) An example for data calculation is shown. Protocol number indicates the corresponding places in the protocol section. Notes explain the meaning of each score. The numerical formula can be referred by clicking the cells. Examples of recovery curve and mobile fraction bar graph are shown. Please click here to download this file.
| Day -3 | Day -2 | Day -1 | Day of FRAP | Day +1 | Day +3 | Day +19 | |
| mRNA preparation | Cutting template plasmid (over night) |
1). Purification template plasmid | |||||
| 2). In vitro transcription | |||||||
| 3). In vitro poly A tailing | |||||||
| 4). mRNA quality check by electrophoresis | |||||||
| FRAP | 1). Preparation of manipulation pipet and chamber | ||||||
| 2). mRNA injection | |||||||
| 3). zFRAP analysis | |||||||
| 4). Calculation of recovery curve and mobile fraction*2 | |||||||
| IVF and analysis of pre-implantation development | eCG injection | 1). hCG injection | 1). Sperm capacitation | ||||
| 2). Pre-incubation of media (HTF) | 2). In vitro fertilization | ||||||
| Preparation of the media (HTF, CZB, H-CZB and PVP-CZB) | 3). Evaluation preimplantation development | ||||||
| Embryo transfer | Preparation of pseudopregnant female (mating with vasectomized male*1) |
Transfer of 2-cell stage embryos to pseudopregnant female | Evaluation of birth rate | ||||
| *1: Vasectomized male mouse should be prepared at least 2 weeks before mating | |||||||
| *2: Calculation can be prolonged into the next day (Day +1) | |||||||
Table 1: Time table of the experiments. The day of the FRAP analysis is regarded as day 0.The experiments that should be performed are indicated on the respective days for each item.
| Categories | No. of injected zygotes | No. of recovered zygotes after mRNA injection (%)* | No. of zygotes analyzed | No. of transferred | No. of recipients | No. of pups (%)*** | Weight of pups (g) | No. of transgenic pups |
| 2-cell stage embryos (%)** | ||||||||
| Intact | – | – | 99 | 98 (99.0) | 9 | 61 (62.2)a | 1.64±0.02 | n.d |
| No FRAP | 420 | 398 (94.8) | 82 | 78 (95.1) | 7 | 41 (52.6)a, b | 1.66±0.03 | n.d |
| FRAP | 79 | 78 (98.7) | 7 | 32 (41.0)b | 1.69±0.03 | 0 | ||
| *: calculated by dividing with no. of injected zygotes; **: calculated by dividing with no. of zygotes analyzed; ***: calculated by dividing with no. of transferred 2-cell stage embryos. a,b: superscripts indicate significant difference (P<0.05). n.d: not determined. This table has been modified from [Ooga et al 2017]7. | ||||||||
Table 2: The birth rate of analyzed embryos: The developmental potential of the embryos, which had been FRAP-analyzed, to full-term was examined. The obtained birth rate of these embryos is shown. As controls, intact and no FRAP-analyzed embryos were also examined. The weights of pups derived from these transferred embryos are shown as well.
