Generation of Human Primordial Germ Cell-like Cells at the Surface of Embryoid Bodies from Primed-pluripotency Induced Pluripotent Stem Cells
Summary
Primordial germ cells (PGCs) are common precursors of both sperm and eggs. Human embryonic PGCs are specified from pluripotent epiblast cells through interactions of cytokines. Here, we describe a 13-day protocol of inducing human cells transcriptomally resembling PGCs at the surface of embryoid bodies from primed-pluripotency induced pluripotent stem cells.
Abstract
Primordial germ cells (PGCs) are common precursors of all germline cells. In mouse embryos, a founding population of ~40 PGCs are induced from pluripotent epiblast cells by orchestrated exposures to cytokines, including bone morphogenetic protein 4 (Bmp4). In human embryos, the earliest PGCs have been identified on the endodermal wall of yolk sac around the end of the 3rd week of gestation, but little is known about the process of human PGC specification and their early development. To circumvent the technical and ethical barriers of studying human embryonic PGCs, surrogate cell culture models have been recently generated from pluripotent stem cells. Here, we describe a 13-day protocol for robust production of human PGC-Like Cells (hPGCLCs). Human induced pluripotent stem cells (hiPSCs) maintained in the primed pluripotency state are incubated in the 4i naïve reprogramming medium for 48 hours, dissociated to single cells, and packed into microwells. Prolonged maintenance of hiPSCs in the naïve pluripotency state causes significant chromosomal aberrations and should be avoided. hiPSCs in the microwells are maintained for an additional 24 hours in the 4i medium to form embryoid bodies (EBs), which are then cultured in low-adherence plasticware under a rocking condition in the hPGCLC induction medium containing a high concentration of recombinant human BMP4. EBs are further cultured for up to 8 days in the rocking, non-adherent condition to obtain maximum yields of hPGCLCs. By immunohistochemistry, hPGCLCs are readily detected as cells strongly expressing OCT4 in almost all EBs exclusively on their surface. When EBs are enzymatically dissociated and subjected to FACS enrichment, hPGCLCs can be collected as CD38+ cells with up to 40-45% yield.
Introduction
Primordial germ cells (PGCs) are common precursors of all germline cells in both sexes. Most of our knowledge on development of PGCs in mammalian embryos has been obtained through studying laboratory mice1,2. At embryonic day 6.0-6.5 of mouse embryos, 6 or similar small numbers of PGC precursors are located in the epiblast, and a founding population of ~40 PGCs are induced from them in a manner dependent on bone morphogenetic proteins Bmp2 and Bmp4 secreted from adjacent cells. The earliest human PGCs so far identified in embryos were on the endodermal wall of yolk sac at around the end of the third week of gestation3. Because this is the same place as migrating PGCs are observed in mouse embryos, it is likely that the observed human PGCs were in the path of migration but not the founding population. However, studies tracing back earlier stages of PGCs or PGC precursors in human embryos have been missing.
Access to human embryonic PGCs is challenging due to both technical and ethical obstacles. To overcome these hurdles, PGC-like cell culture models have been recently generated from human pluripotent stem cells (PSCs). Pluripotency is the cellular capability to differentiate into the germline and three embryonic germ layers4. Whereas human PSCs maintained in the mTeSR1 medium (a ready-to-use, commercially available medium formulated for maintenance of human PSCs in the primed pluripotency state) on dishes coated with the extracellular matrix protein have primed-state pluripotency4, in 2013 Jacob Hanna's lab showed that the primed pluripotency cells can be converted into a naïve pluripotency state by exposing to the naïve human stem cell medium (NHSM) containing chemical inhibitors to protein kinases ERK1/2, GSK3, JNK, ROCK, PKC, and p38 MAPK as well as growth factors LIF, TGF, bFGF5. From the naïve-pluripotency human PSCs, in 2015 a research group led by Hanna and Azim Surani accomplished the first robust production of human PGC-Like Cells (hPGCLCs) from PSCs6. Later, several other laboratories, including ours, reported generation of hPGCLCs from PSCs using slightly different protocols7,8,9,10. Our study provided evidence that hPGCLCs generated using different protocols (which are summarized in Table S1 of our previously published study10) are transcriptomally similar to each other10. Available evidence supports the resemblance of human PGCLCs to early-stage human embryonic PGCs prior to the global epigenetic erasure7 and/or chemotactic migration10.
Studies of mouse embryonic PGCs, mouse PGCLCs, and human PGCLCs (but with only very limited access to human PGCs) have revealed that molecular mechanisms of PGC specification differ significantly between mouse and human1,6,7,8,9,10,11,12,13,14,15,16,17. For example, Prdm14 plays critical roles in PGC specification in mouse embryos, but its role in human PGC specification seems limited1,15. In contrast, induction of SOX17 by EOMESODERMIN is essential for PGC specification6,11,14, whereas these transcription factors seem dispensable for mouse PGC specification15. These initial achievements of studies using hPGCLCs strongly support the importance of this cell culture model as a surrogate of human embryonic PGCs.
Recently published studies, involving our lab's deep sequencing evaluation of genomic DNA copy number analysis, have shown that prolonged maintenance of PSCs in the naïve pluripotency state significantly increases the risk of chromosomal instability and structural anomalies. This phenomenon was observed with both mouse18 and human19 PSCs. The original hPGCLC production protocol reported by Hanna/Surani was developed for human PSCs maintained in the 4i naïve pluripotency medium for at least 2 weeks6. To preserve the normal diploid karyotype of human PSCs and PGCLCs, we developed a modified protocol in which human PSCs are exposed to the 4i medium for only 72 hours10, which is presented in this article. Human iPSCs (hiPSCs) are maintained under the primed pluripotency state. Immediately before EB formation, cells are incubated in the 4i naïve reprogramming medium (a modified NHSM medium) for 48 hours. Cells are then dissociated and packed into microwells to form EBs for an additional 24 hours in the 4i medium. EBs are maintained in hPGCLC induction medium containing a high concentration of recombinant human BMP4 under a rocking condition for no-attachment culture for up to 8 days to obtain the maximum yield of hPGCLCs. After 8-day EB culture, hPGCLCs can be isolated from dissociated EB cells by FACS as CD38+ cells with up to ~40% yield in FACS-sortable single cell suspension. Whereas other published methods7,8,9, including the original protocol before our modifications6, typically generate hPGCLCs in spontaneously formed cell aggregates without specific localization, hPGCLC produced by our protocol are observed at the surface of embryoid bodies (EBs).
Protocol
1. Cell Culture of hiPSCs in the Primed Pluripotency State
- Preparation of extracellular matrix protein-coated dishes
- Thaw Matrigel (extracellular matrix protein) on ice in a refrigerator or a cold room overnight (do not thaw at room temperature or using a warm water bath). Aliquot matrix protein (~200 μL; the volume of an aliquot is determined by manufacturer for each batch and indicated on the label of vial) into sterile low-bind centrifuge tubes or cell cryopreservation tubes on ice. It is important to keep extracellular matrix protein ice-cold during dispensing to avoid solidification. Store the aliquots at -80 °C.
- To coat cell culture plastic dishes with extracellular matrix protein, dilute one aliquot with 25 mL ice-cold DMEM/F12 and spread to three 10-cm dishes (~8 mL/dish). Incubate dishes at room temperature for at least 1 hour. After coating, dishes can be sealed using Parafilm and stored at room temperature for up to one week.
- Aspirate medium from dishes immediately before inoculation of primed pluripotency hiPSCs. It is not necessary to wash the coated dishes with medium or calcium/magnesium-free phosphate-buffered saline [PBS(-)].
- Initiation of hiPSC culture in the primed pluripotency state
- Add 2 µL of 50 mM Y27632 [inhibitor of Rho-associated protein kinase (ROCK)] to 10 mL of mTeSR1 medium in a 15-mL centrifuge tube. Prepare two tubes of ROCK inhibitor-supplemented 10 mL medium to initiate cell culture in one 10-cm dish from 1 mL frozen cell stock. Use Y27632-supplemented medium on the same day. Prewarm Y27632-supplemented medium in a 37 °C water bath for 5 min.
Note: This protocol works well with hiPSCs maintained in mTeSR1. When hiPSCs are maintained in other media supporting human PSC growth with varying degrees of primed or naïve pluripotency, yield of hPGCLCs may significantly vary.
Note: The shelf life of complete mTeSR1 is 2 weeks at 4 °C and 6 months at -20 °C, but re-freezing causes poor performance. We freeze 40 mL aliquots of mTeSR1 at -20 °C until use. - Thaw a vial of primed pluripotency hiPSC frozen stock (1.0 – 3.0 x 106 cells in 1 mL cryopreservation medium) using a 37 °C water bath. Immediately after completion of thawing, transfer the entire content of a vial into one tube (10 mL) of prewarmed Y27632-supplemented medium.
- Centrifuge cell suspension at room temperature, 300 x g for 8 min. Discard supernatant and resuspend cell pellet with 10 mL prewarmed Y27632-supplemented from the other tube.
- Evenly spread cell suspension into an extracellular matrix protein-coated 10-cm dish. Place the dish in a tri-gas CO2 incubator (37 °C, 6.5% O2, 5% CO2). Maintain hiPSC culture under a low oxygen pressure.
- Change medium (mTeSR1 without Y27632) every day. The ROCK inhibitor (Y27632) is critical for hiPSC survival immediately after dissociation to single cells. Once hiPSCs adhere on the extracellular matrix protein-coated surface as evenly distributed small aggregates with >10% confluency (which is typically completed within 16 hours after inoculation), Y27632 is dispensable. Too early medium change after inoculation of dissociated hiPSCs without Y27632 may cause almost complete cell death.
- Add 2 µL of 50 mM Y27632 [inhibitor of Rho-associated protein kinase (ROCK)] to 10 mL of mTeSR1 medium in a 15-mL centrifuge tube. Prepare two tubes of ROCK inhibitor-supplemented 10 mL medium to initiate cell culture in one 10-cm dish from 1 mL frozen cell stock. Use Y27632-supplemented medium on the same day. Prewarm Y27632-supplemented medium in a 37 °C water bath for 5 min.
- Passaging primed pluripotency hiPSCs
Note: Passage primed pluripotency hiPSCs when colonies occupy ~80% of the effective growth area. Correct passaging procedures and densities is important for experimental reproducibility. We have seen that efficiency of hPGCLC induction is not significantly different between hiPSC cultures 6 days after initiation from a frozen stock (involving one or two passages) or one month (involving >10 passages). Induction of hPGCLC was successfully performed from hiPSCs maintained for over two months, although the efficiency was not quantitatively assessed.- Take 4 mL of cell dissociation enzyme mixture in a 15-mL centrifuge tube and equilibrate to room temperature for 5 min.
- Prewarm 10 mL PBS(-) in a 15-mL centrifuge tube for >5 min at 37 °C in a water bath.
- Add 2 µL of 50 mM Y27632 (ROCK inhibitor) to 10 mL mTeSR1 medium in a 15-mL centrifuge tube. Prepare two tubes of the ROCK inhibitor-supplemented 10 mL medium and use in the same day. In another 15-mL centrifuge tube, take 5 mL mTeSR1 without adding Y27632. Prewarm medium +/- Y27632 in a 37 °C water bath for 5 min.
Note: All medium aliquots prepared in step 1.3.3 may contain Y27632. - Aspirate old medium from a 10-cm dish and rinse cells once with prewarmed 10 mL PBS(-). Discard PBS(-) and add 4 mL of dissociation enzyme to the dish. Place the dish in a CO2 incubator (tri-gas incubator works but not necessary) for ~4 min until cells detach from the bottom. Gently pipet cells up and down to make single-cell suspension.
- Transfer hiPSC single-cell suspension to the prewarmed tube containing 5 mL medium without Y27632 (total volume in a 15-mL tube is about 9 mL). Centrifuge cell suspension at room temperature, 300 x g for 8 min.
- Discard supernatant and resuspend cell pellet with 10 mL of the prewarmed medium medium containing Y27632.
- Remove cell aggregates using a 40-µm cell strainer and determine cell density using a Coulter counter.
- Dilute cell suspension with prewarmed Y27632-supplemented medium to achieve 3.0 x 106 cells in 10 mL to inoculate an extracellular matrix protein-coated 10-cm dish. Place the inoculated dish in a tri-gas CO2 incubator (37 °C, 6.5% O2, 5% CO2).
- Change medium (mTeSR1 without Y27632) every day.
2. Generation of hPGCLCs
Note: Initiate the following steps when hiPSC cells in a 10 cm dish (mTeSR1 medium, extracellular matrix protein-coated) reaches to ~80% confluency (approximately 107 cells).
- Preparation of extracellular matrix protein-coated dishes (Day 1)
- Thaw one aliquot of extracellular matrix protein on ice and dilute in 25 mL of ice-cold DMEM/F12.
- Dispense diluted extracellular matrix protein to 6-well plates (1 mL/well). One aliquot is sufficient to coat 24 wells (4 plates). Incubate plates at room temperature for at least 1 hour. Unused coated plates can be sealed and stored at room temperature for up to one week.
- Aspirate medium from plates immediately before inoculation of primed pluripotency hiPSCs. It is not necessary to wash the coated dishes with medium or PBS(-).
- Inoculation of primed pluripotency hiPSCs into extracellular matrix protein-coated plates (Day 1)
- Take 4 mL of cell dissociation enzyme in a 15-mL centrifuge tube and equilibrate to room temperature for 5 min.
- Prewarm 10 mL PBS(-) in a 15-mL centrifuge tube for >5 min at 37 °C in a water bath.
- Add 7 µL of 50 mM Y27632 (ROCK inhibitor) to 35 mL mTeSR1 medium in a 50-mL centrifuge tube. Use Y27632-supplemented medium in the same day. Make two tubes for total 70 mL Y27632-supplemented medium and prewarm the medium in a 37 °C water bath for 15 min.
- Aspirate old medium from a 10-cm dish and rinse cells once with prewarmed 10 mL PBS(-). Discard PBS(-) and add 4 mL of cell dissociation enzyme to the dish. Place the dish in a CO2 incubator (tri-gas incubator works but not necessary) for ~4 min until cells detach from the bottom. Gently pipet cells up and down to make single-cell suspension.
- Transfer hiPSC single-cell suspension to the prewarmed tube containing 10 mL Y27632-supplemented medium. Centrifuge cell suspension at room temperature, 300 x g for 8 min.
- Discard supernatant and resuspend cell pellet with 10 mL prewarmed Y27632-supplemented medium.
- Filter cell suspension through a cell strainer (40 µm pore size) to remove cell aggregates and count cells using a Coulter counter.
- Dilute hiPSC single cell suspension to 5.0 x 106 cells in 50 mL prewarmed Y27632-supplemented medium.
- Inoculate 2 mL cell suspension (2.0 x 105 cells) in each well of the coated 6-well plates (4 plates, 24 wells). Make sure that cells are evenly distributed in each well. Place cell culture plates in a tri-gas CO2 incubator (37 °C, 6.5% O2, 5% CO2).
Note: The inoculum cell density and even distribution in each well is critical. Because inoculation of hiPSCs into 10-cm dishes may cause significantly higher cell density than other areas. Even cell density of single-cell hiPSCs can be achieved more easily with 6-well plates. - Change medium with 2 mL/well mTeSR1 (without Y27632) at 24 hours after inoculation.
- Conversion of the primed pluripotency state hiPSCs to the 4i-naïve (ERK-independent) pluripotent cells (Day 3 and Day 4)
- Prepare 4i complete naïve pluripotency medium in a 50-mL centrifuge tube as follows (Day 3 and Day 4): 50 mL of 4i basal medium, 5 µL of 30 mM CHIR99021, 5 µL of 10 mM PD0325901, 5 µL of 20 mM BIRB796, 5 µL of 50 mM SP600125, and 5 µL of 50 mM Y27632.
Note: Store 4i basal medium at -80 °C and thaw in a refrigerator overnight. Prepare fresh 4i complete medium immediately before use. Avoid exposure of the 4i chemicals (CHIR, PD, BIRB, and SP) to strong light. Do not store 4i complete medium at 4 °C or frozen overnight or longer. - Warm 50 mL 4i complete medium in a 37 °C water batch for 15 min. Change medium with prewarmed 4i complete medium (2 mL/well) at 48 and 72 hours after inoculation. Exact timing of medium change is important to achieve high hPGCLC yield and experimental reproducibility.
Note: Density of the 4i hiPSC culture is high under this condition and become confluent at 48-72 hours after inoculation, but this is normal.
- Prepare 4i complete naïve pluripotency medium in a 50-mL centrifuge tube as follows (Day 3 and Day 4): 50 mL of 4i basal medium, 5 µL of 30 mM CHIR99021, 5 µL of 10 mM PD0325901, 5 µL of 20 mM BIRB796, 5 µL of 50 mM SP600125, and 5 µL of 50 mM Y27632.
- EB formation using microwell plates (Day 5)
- Prepare 50 mL of 4i complete medium in a 50-mL centrifuge tube and prewarm it in a 37 °C water bath for ~15 min before use.
- Prewarm 35 mL DMEM/F12 in a 50-mL centrifuge tube in a 37 °C water bath for ~15 min before use.
- Prepare a microwell plate
- Add 5% (w/v) filter-sterilized Pluronic F-127 detergent into 8 active wells of a microwell plate (see Table of Materials; 0.5 mL/well).
- Centrifuge the microwell plate at room temperature, 1,000 x g, for 5 min. Inspect microwells with an inverted microscope to ensure the absence of air bubbles. Leave the microwell plate for 30 min at room temperature to coat microwells with detergent.
- Discard detergent solution from wells of the microwell plate by pipetting and rinse wells with prewarmed DMEM/F12 (2 mL/well).
- Centrifuge the microwell plate at room temperature, 1,000 x g for 5 min. Inspect microwells with an inverted microscope to ensure the absence of air bubbles.
Note: When air bubbles are still observed in microwells, examine if the microwell sheet is still firmly glued to the bottom of the well. - Discard DMEM/F12 from wells of the microwell plate by pipetting and rinse wells with prewarmed DMEM/F12 (2 mL/well).
- Repeat steps 2.4.3.3 – 2.4.3.4.
- Add 4i complete medium into the rinsed wells of the microwell plate (1 mL/well). Keep the remaining 4i complete medium in a 37 °C water bath.
- Centrifuge the microwell plate at room temperature, 1,000 x g for 5 min. Inspect microwells with an inverted microscope to ensure the absence of air bubbles.
- Place the microwell plate in a tri-gas incubator (37 °C, 6.5% O2, 5% CO2) until inoculation of 4i hiPSCs.
- Inoculation of 4i iPSCs into a microwell plate
- Take 13 mL of cell dissociation enzyme in a 15-mL centrifuge tube and equilibrate to room temperature for 5 min.
- Prewarm 50 mL PBS(-) in a 50-mL centrifuge tube for >5 min at 37 °C in a water bath.
- Aspirate old medium from 24 wells of naïve hiPSC cell culture and rinse cells once with prewarmed 2 mL/well PBS(-). Discard PBS(-) and add 0.5 mL/well dissociation enzyme. Place cell culture plates in a CO2 incubator (37 °C) for ~4 min until cells detach from the bottom. Gently pipet cells up and down to make single-cell suspension.
- Transfer hiPSC single-cell suspension to prewarmed 20 mL 4i complete medium. Centrifuge cell suspension at room temperature, 300 x g for 8 min.
- Discard supernatant and resuspend cell pellet with 5 mL prewarmed 4i complete medium.
- Filter cell suspension through a cell strainer (40 µm pore size) to remove cell aggregates. Wash cell strainer 3 times with 1 mL prewarmed 4i complete medium. Count cells using a Coulter counter.
- Dilute single cell suspension of 4i naïve hiPSCs to 27.0-32.4 x 106 cells in 9 mL prewarmed 4i complete medium. Note that 4i complete medium already contains Y27632.
- Take the microwell plate containing 1 mL of 4i complete medium in 8 well from a tri-gas incubator (2.4.3.9). Inoculate 1 mL 4i naïve hiPSC suspension (3.0-3.6 x 106 cells/well) into each well. This cell density is critical. Make sure that cells are evenly distributed in each well by gently pipetting.
- Centrifuge the microwell plate at room temperature, 100 x g for 3 min.
- Place the microwell plate in a tri-gas CO2 incubator (37 °C, 6.5% O2, 5% CO2). Do not disturb cells pelleted in microwells. Incubate cells for 24-30 hours. An overnight incubation (~16 hours) is typically insufficient for formation of tight EBs.
- Transfer of EBs to low attachment plates for rocking culture (Day 6)
- Prepare hPGCLC complete medium in a 50 mL centrifuge tube as follows (Day 6 – Day 13):
20 mL hPGCLC basal medium, 4 μL of 500 mM 2-Mercaptoethanol, 50 μL of 20 mg/mL L-Ascorbic acid, 4 μL of 50 mM Y27632, 50 μL of 100 μg/mL recombinant human BMP4, 4 μL of 500 μg/mL human SCF, 4 μL of 250 μg/mL human EGF, and 8 μL of 250 μg/mL human LIF.
Note: Prepare fresh hPGCLC complete medium immediately before use and avoid exposure to light. Do not store hPGCLC complete medium at 4 °C or frozen overnight or longer.
Note: The concentration of BMP4 is very high. The optimal BMP4 concentration may differ between batches of BMP4. The lot-to-lot difference of BMP4 strongly affect hPGCLC production. In the absence of BMP4 in the complete hPGCLC medium, yield of hPGCLCs is very low6. The importance of other cytokine in the complete hPGCLC medium was described by Hanna/Surani6.
Note: In this protocol, the final concentration of human LIF in the complete hPGCLC medium is 100 ng/mL6,10. The final LIF concentration used in previously published studies, including ours, was 1 μg/mL. When the specific activity of human LIF reagent is >10,000 units/μg (ED50 < 0.1 ng/mL), 100 ng/mL LIF is sufficient to support the hPGCLC generation from EBs. - Transferring EBs
- Prewarm 5 mL hPGCLC basal medium and 20 mL hPGCLC complete medium for >5 min at 37 °C in a water bath.
- Carefully place a cell strainer upside down atop a 50 mL polypropylene conical tube.
- Pipet each well of the microwell plate very gently plate to detach EBs from microwells. Filter all contents of each well through the cell strainer to remove cells not incorporated in EBs.
- Wash EBs retained on the cell strainer with 1 mL of prewarmed hPGCLC basal medium. Gently repeat wash 5 times.
- Washed EBs are now retained on the membrane of a strainer, which is on a 50-mL conical tube upside-down. Place a fresh 50-mL conical tube on the cell strainer so that the cell strainer is normally positioned in the new tube. Then quickly invert the cell strainer with the new tube. The cell strainer and the tube are now in the normal positions, and EBs are below the membrane of the cell strainer.
- Collect EBs in the conical tube by adding 18 mL prewarmed hPGCLC complete medium from above the membrane of cell strainer. Thus, medium will go through the membrane and collect EBs attached on the lower side of the membrane down to the bottom of the centrifuge tube.
- Plate suspension of EBs into wells of a low-attachment 6-well plate (3 mL/well).
- Place the low-attachment plate on a seesaw-move rocker in a tri-gas incubator (37 °C, 6.5% O2, 5% CO2). Set the rocking speed at ~20 turns per min.
Note: Do not apply swirling movement because EBs aggregate at the center of wells and fuse. Carefully adjust rocking speed so that EBs do not aggregate. Too vigorous rocking will physically damage EBs.
Note: Low oxygen pressure is strongly recommended for EB culture.
- Prepare hPGCLC complete medium in a 50 mL centrifuge tube as follows (Day 6 – Day 13):
- Generation of hPGCLCs on the surface of EBs (Day 7 – Day 13)
- Freshly prepare 20 mL hPGCLC complete medium each day. Without rocking, EBs will sink down on the bottom of wells in ~1 min. Remove old medium without drying EBs (~0.2 mL/well old medium may remain) and add fresh hPGCLC complete medium every day (3 mL/well).
3. Immunohistochemical staining of EBs (Day 10 – Day 13)
- Embedding EBs in extracellular matrix protein blocks for formaldehyde-fixed, paraffin-embedded (FFPE) immunostaining
- To identify hPGCLCs as OCT4+ cells, harvest EBs in a 1.5 mL low-bind microcentrifuge tube. Let EBs sink to the bottom or briefly centrifuge (2 seconds) at room temperature and discard medium. Rinse EBs with ice-cold PBS(-).
- Remove PBS(-) and incubate EBs in 1 mL of ice-cold 1%(w/v) sodium azide in PBS(-) for 5 min on ice. Let EBs sink to the bottom or briefly centrifuge (2 seconds) at room temperature. Discard supernatant, and rinse EBs with ice-cold PBS(-).
Note: Addition of sodium azide slows down degradation of intracellular proteins and RNA transcripts. - Thaw 200 µL of extracellular matrix protein on ice and add to the microcentrifuge tube containing EBs. Leave the tube at room temperature for ~10 min until the gel is solidified.
- Add 1 mL of 4% formaldehyde in PBS(-) and incubate at room temperature for 15 min with gentle rocking.
Note: Both EBs and extracellular matrix protein are fixed during this step. Without fixation, the gel blocks may decompose during the process of dehydration. If pre-fixed EBs are embedded in the gel block, EBs are fixed again with the gel block and can be over-fixed. - Discard formaldehyde, and rinse EBs once with ice-cold PBS(-). Add 1 mL of 70% ethanol. Gel-embedded EBs can be stored in 70% ethanol at 4 °C for one week.
- Process the gel-embedded EBs with a standard FFPE protocol. Prepare 5-μm thickness FFPE slides. Let slides dry overnight and store them at room temperature until immunostaining.
Note: Our routine FFPE protocol is as follows: 70% ethanol, two changes, 1 hour each; 80% ethanol, one change, 1 hour; 95% ethanol, one change, 1 hour; 100% ethanol, three changes, 1.5 hour each; xylene, three changes, 1.5 hour each; paraffin wax (58-60 °C), two changes, 2 hours each. The dehydrated tissues are embedded into paraffin blocks and cut at 3-10 μm (typically 5 μm). - For staining, completely dry slides in 56 °C oven for at least 30 min. Deparaffinize and hydrate slides with xylenes and graded alcohol series. Then wash slides in tap water for 5 min.
- To inactivate endogenous peroxidases, incubate rehydrated slides with BLOXALL Blocking Solution at room temperature for 10 min. Then wash slides with PBS for 5 min.
- Block slides with Normal Horse Serum (or normal sera of other species generated secondary antibodies) at room temperature for 20 min.
- Incubate slides with an anti-OCT-3/4 goat primary antibody diluted with 0.1% BSA in PBS(-) at 4 °C overnight (optimize dilution and incubation conditions for each primary antibody). Then wash slides with PBS(-) three times at room temperature, 5 min each.
- Incubate slides with the Anti-Goat IgG (horseradish peroxidase-conjugated secondary antibody generated by horse; see Table of Materials) at room temperature for 30 min. Then wash slides with PBS(-) three times at room temperature, 5 min each.
- Mix necessary amounts the DAB staining substrates (see Table of Materials) immediately before use and incubate slides in complete DAB staining solution at room temperature for 5 min. Monitor DAB staining using an inverted microscope and stop the reaction when OCT4+ cells are visualized by quickly rinsing slides in flowing tap water.
- Dehydrate the slides with graded alcohol and xylenes series. The slides are ready for laser-capture microdissection. Prepare permanent FFPE slides using mounting medium (see Table of Materials) for high-resolution microscopic observations.
4. FACS enrichment of hPGCLCs from EBs (Day 10 – Day 13)
- Preparing enzyme mix of Embryoid Body Dissociation Kit
- Prepare Enzyme Mix 1 by adding 50 µL Enzyme P to 1,900 µL of Buffer X and vortex. Prewarm Enzyme Mix 1 in a 37 °C water bath for 15 min.
- Prepare Enzyme Mix 2 by adding 10 µL Enzyme A to 20 µL Buffer Y.
- Mix Enzyme Mixes 1 and 2 to prepare Complete EB Dissociation Enzyme Mix.
- Dissociating EBs
- Harvest EBs from low attachment plates and centrifuge at room temperature in a 15-mL centrifuge tube, 300 x g for 2 min. Discard supernatant and resuspend EBs with 10 mL PBS(-). Centrifuge again.
- Discard supernatant and resuspend EBs with Complete EB Dissociation Enzyme Mix (4.1.3). Incubate EB suspension in a 37 °C water bath for 15 – 20 min until EBs are dissociated. During incubation, gently pipet EBs at every 3 – 5 min to facilitate dissociation. Alternatively, use an automated dissociator with Embryoid Body Dissociation Program.
- Add ice-cold 8 mL PBS(-) to dissociated EB cell suspension. Filter the cell suspension through a 40-µm cell strainer. Wash cell strainer with 1 mL ice-cold PBS(-) three times.
- Count cells in filtered cell suspension using a Coulter counter.
- Centrifuge cell suspension at at 4 °C, 300 x g for 8 min and discard supernatant. Resuspend cell pellet with ice-cold 10 mL PBS(-).
- Divide cell suspension into two tubes, one for no-staining control (~105 cells) and the other for anti-CD38 staining (all remaining cells).
- Centrifuge the two tubes at 4 °C, 300 x g for 8 min.
- Anti-CD38 staining and FACS enrichment of hPGCLCs
- Resuspend cell pellet for no-staining control with 200 µL ice-cold 3% (w/v) BSA and 1% (w/v) sodium azide in PBS(-). Place on ice until flow cytometry.
Note: Addition of sodium azide slows down internalization of CD38 from cell surface. - Resuspend cell pellet for anti-CD38 staining with ice-cold 3% (w/v) BSA and 1% (w/v) sodium azide in PBS(-) to 1 x 106 cells per 100 µL.
- Add 10 µL per 1 x 106 cells of a FACS-grade, APC-conjugated anti-CD38 antibody. Cover the tube with aluminum foil to avoid exposure to light. Incubate at 4 °C for 45 min with gentle rocking.
- Add 5 mL ice-cold PBS(-) and centrifuge at 4 °C, 300 x g for 8 min. Discard supernatant and resuspend cell pellet with 5 mL ice-cold PBS(-). Repeat wash with PBS(-) three times in total.
- Resuspend cell pellet with 500 μL ice-cold 3% (w/v) BSA and 1% (w/v) sodium azide in PBS(-) to a cell density adequate for FACS.
- Enrich hPGCLCs as CD38+ cells, which typically form clearly distinguished cell population. Use the no-staining control to ensure identification of CD38+ cells. Typical yield of hPGCLCs are 1-5% of all FACS-examined single cells from Day 10 EBs and 20-40% from Day 13 EBs.
- Resuspend cell pellet for no-staining control with 200 µL ice-cold 3% (w/v) BSA and 1% (w/v) sodium azide in PBS(-). Place on ice until flow cytometry.
Representative Results
The microwell plate used here is in the 24-well format and has 8 wells holding the microwell sheets, each of which supports formation of up to 1,200 EBs. From approximately 24 million of 4i naïve pluripotency cells, this microwell plate typically generates ~8,000 EBs consisting of ~3,000 cells per EB. During non-adherent culture of EBs with constant rocking, the number of intact EBs gradually decreases due to spontaneous self-dismantling, and ~3,000 EBs survive until Day 13 of the protocol. Most of these surviving EBs have 50-200 hPGCLCs on their surface (estimated by immunohistochemical detection of OCT4+ cells in serial sections of EBs; see Figure 8), yielding ~100,000 OCT4+ hPGCLCs in total. Enzymatic digestion of EBs into single cells is a relatively inefficient process, reducing the yield of FACS-enriched CD38+ hPGCLCs to 9,000 – 47,000 cells. In our hands, an average of six independent but consecutive batches of experiments was 14,038 ± 5,731 (mean ± SEM). Because CD38-negative EB cells cells express OCT4 mRNA (qPCR) or protein (Immunohistochemistry) only very weakly, if not completely absent, all EB cells strongly expressing OCT4 protein are practically equivalent to the whole population of the CD38+ hPGCLCs.
The critical parameter of this protocol includes cell density. When 2.0 x 105 human iPSCs are inoculated in an extracellular matrix protein-coated well of 6-well cell culture plate (9.60 cm2 growth area per well) with 2 mL Y27632-supplemented medium (2.2.9), cells will reach to about 20-30% confluency at 24 hours after inoculation (Figure 1). After an additional 24-hour culture in the mTeSR1 medium in the absence of Y7632, cells aggregate and form colonies, occupying ~30% of the growth area (Figure 2). Cells are then cultured in the 4i reprogramming medium (2.3.2). After 24 hours of culture in the 4i medium, cells become confluent (Figure 3). An additional 24-hour culture in the 4i medium makes cells densely packed (Figure 4). The exact timing of medium change (every 24 hours +/- 4 hours) and cell densities are critical for successful formation of EBs and hPGCLCs.
After 48-hour culture in the 4i medium, cells are dissociated and inoculated to the microwell plate, which has wells 400 µm in size (2.4). Although this commercially available microwell plate is coated for low-adhesion surface by the manufacturer, fresh re-coating with detergent (2.4.3) is recommended to reduce the risk of unwanted cell adhesion. 800 µm microwells resulted in reduced yield of hPGCLCs, suggesting the importance of EB size for properly directed differentiation. Cells inoculated in the 4i medium will form EBs in microwells at 24-30 hours, which can be observed using a standard, inverted phase contrast microscope (Figure 5). The circular contour of EBs become clearly visible at and after 24 hours of incubation. Harvesting EBs at an earlier time (e.g., 16 hours after inoculation) before their circular contour is clearly visible is not recommended because such EBs are very vulnerable to mechanistic damages and easily dismantle. Note that significant amounts of naïve hiPSCs are NOT incorporated in EBs, which is normal. These unincorporated cells will be washed away before initiation of rocking culture of EBs on low-adherent surface (2.5.2). Also note that, in our protocol, EBs are formed in the 4i naïve pluripotency medium – not in the hPGCLC medium. Pre-formation of solid EBs in the 4i medium before exposure to the hPGCLC medium is important for distribution of hPGCLCs on the surface of EBs.
EBs maintained in the hPGCLC medium under a rocking, non-adherent culture condition will maintain their spherical shape without aggregation or fusion (Figure 6 and Figure 7). Too weak rocking condition will cause EB aggregation and fusion, but too harsh condition will dismantle EBs. Human PGCLCs emerge as OCT4-expressing cells on the surface of EBs after as early as the 5-day culture in the hPGCLC medium, and their number increases until the 8-day culture (Figure 8). Further incubation of EBs may cause dismantling of EBs and loss of hPGCLCs. Human PGCLCs can be enriched from enzymatically dissociated EB cells after 5-8 days of culture in the hPGCLC medium by FACS as CD38+ cells (Figure 9).
Figure 1: Human iPSC cell culture in Y27632-supplemented medium 24 hours after inoculation. Cell density is about 20-30% confluent. In the presence of ROCK inhibitor Y27632, cells tend to spread well with long, spike-like elongation. Scale bar = 100 μm. Please click here to view a larger version of this figure.
Figure 2: Human iPSC cell culture 48 hours after inoculation. Cells aggregate to form colonies, occupying ~30% of growth area. Scale bar = 100 μm. Please click here to view a larger version of this figure.
Figure 3: Human iPSC cell culture incubated in the 4i reprogramming medium for 24 hours. Cells reach to confluency. Scale bar = 100 μm. Please click here to view a larger version of this figure.
Figure 4: Human iPSC cell culture incubated in the 4i reprogramming medium for 48 hours. Confluent cells are densely packed. Scale bar = 100 μm. Please click here to view a larger version of this figure.

Figure 5: Human iPSC EBs formed in microwells after 24-hour incubation in the 4i reprogramming medium. The circular contour of EBs become visible at 24-30 hours after inoculation. When the contour is confirmed under a phase contrast microscope, EBs are ready for transfer to a rocking culture condition. Scale bar = 500 μm. Please click here to view a larger version of this figure.
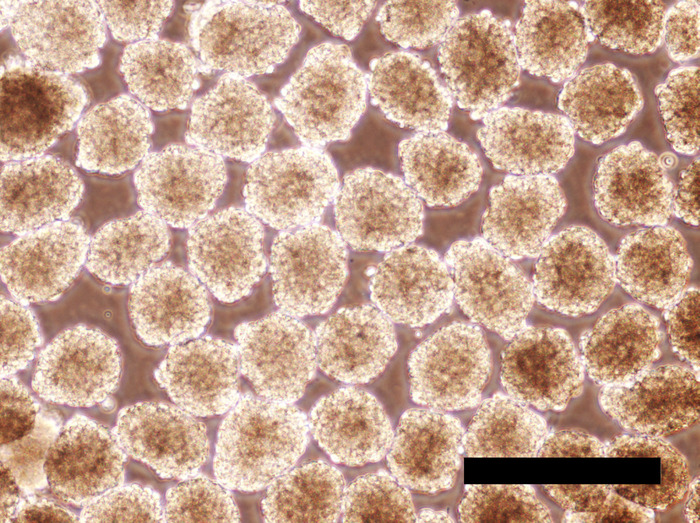
Figure 6: Human iPSC EBs incubated for 24 hours in the hPGCLC medium. EBs largely maintain their spherical shape. Scale bar = 500 μm. Please click here to view a larger version of this figure.
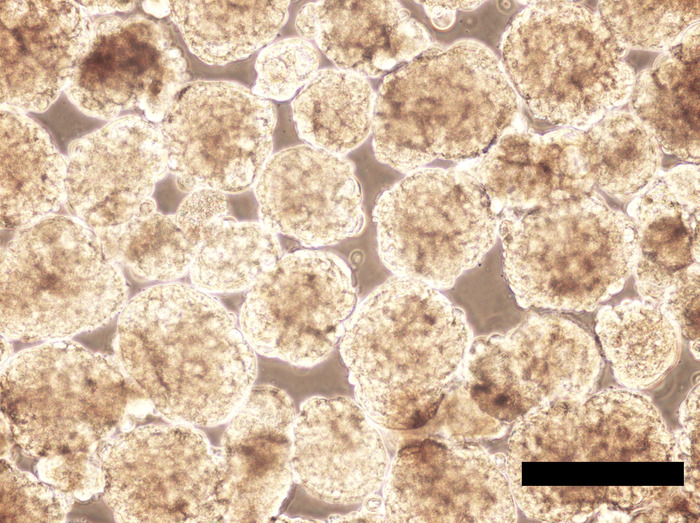
Figure 7: Human iPSC EBs incubated for 192 hours in the hPGCLC medium. EBs are enlarged compared to their appearance at 24-hour culture, but they still largely maintain spherical shapes with no aggregation or fusion. Scale bar = 500 μm. Please click here to view a larger version of this figure.
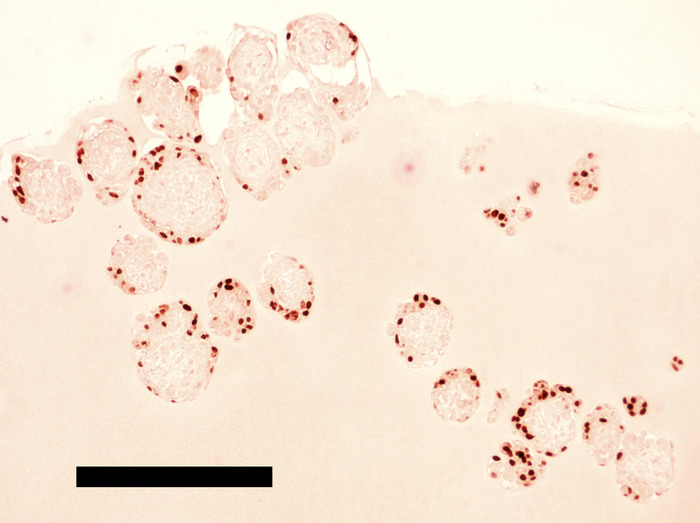
Figure 8: Human PGCLCs expressing OCT4 are localized on the surface of hiPSC EBs incubated for 192 hours in the hPGCLC medium. EBs were embedded in extracellular matrix protein and processed for FFPE slide immunohistochemical staining of OCT4 (DAB substrate). Scale bar = 1 mm. Please click here to view a larger version of this figure.
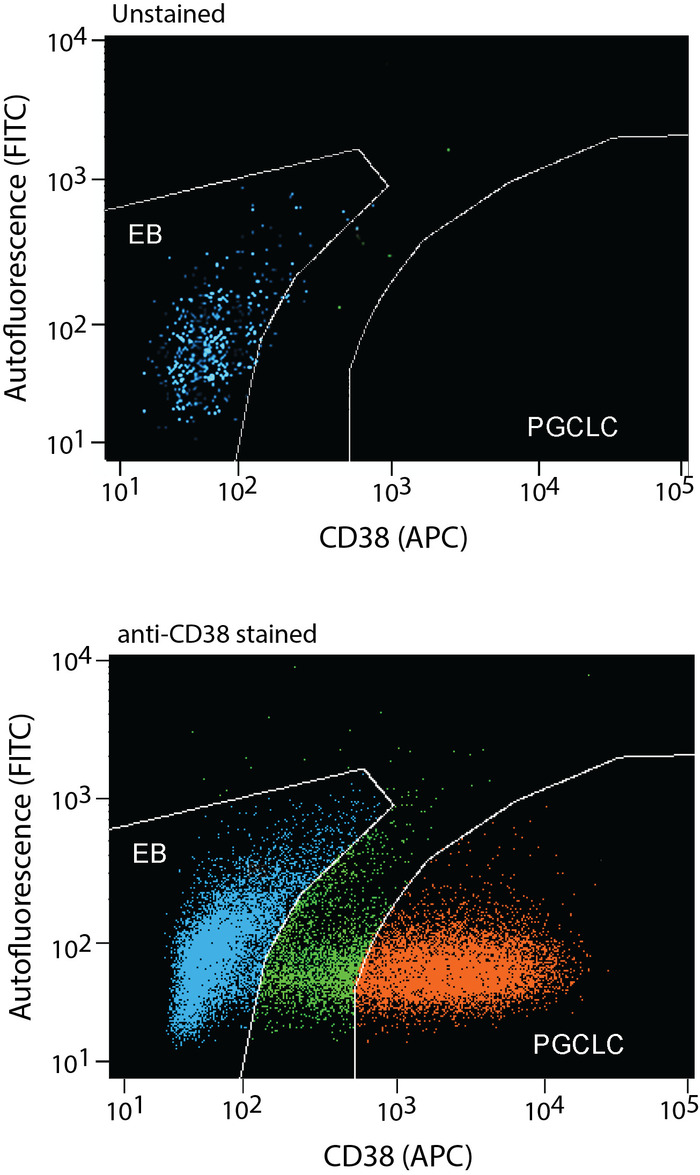
Figure 9: Human PGCLCs are enriched from enzymatically dissociated EB cells by FACS as CD38+ cells. After incubation in the hPGCLC medium for 5-8 days, EBs can be dissociated by enzymatic digestion to prepare single cell suspension. hPGCLCs can be enriched as CD38+ cells by FACS (red dots). EB cells that do not express CD38 (blue dots) should also be collected as negative control. FACS gates of CD38-positive and CD38-negative cells should be separated with a wide margin (green dots) to avoid contamination of each type of cells. The upper and lower panels show FACS profiles without or with anti-CD38 antibody staining, respectively. Please click here to view a larger version of this figure.
Discussion
Robust production of hPGCLCs using the protocol described here was confirmed with three independent clones of human iPSCs with the normal diploid karyotype10. These iPSC clones were derived from the same human neonatal dermal skin fibroblast cell culture10. They will be provided by authors of this article to investigators upon request and under appropriate materials transfer agreement and shipping arrangement of frozen live human cells. It is presently unknown as to whether normal karyotype is required for robust hPGCLC production using our protocol or those reported by other laboratories.
Recent studies have shown that production of hPGCLCs from hiPSCs11 or ESCs14 using a protocol described by Saitou's group of Kyoto University8 is dependent on expression of EOMESODERMIN, a T-box transcription factor required for induction of SOX17. SOX17 seems to function as the master lineage determining transcription factor in germline differentiation of human pluripotent stem cells6. EOMESODERMIN is encoded by the EOMES gene, and CRISPR/Cas9 knockout of EOMES caused almost complete absence of SOX17 induction in the hPGCLC producing condition11, and expression of other genes followed the same pattern of the SOX17-null knockout cells. Overexpression of EOMESODERMIN from an inducible vector in the EOMES-null knockout cells during hPGCLC induction culture efficiently rescued the robust hPGCLC production as well as induction of germline genes, including SOX17. In contrast, induced overexpression SOX17 also rescued the robust PGCLC production but without inducing EOMES. Thus, EOMESODERMIN is a critical upstream inducer of SOX17, and this seems the single most important role of EOMESODERMIN in hPGCLC induction from human pluripotent stem cells. Our protocol induces SOX17 in hiPSCs10, but its dependency on EOMESODERMINE induction awaits to be determined.
This protocol converts primed-pluripotency human iPSCs maintained in the mTeSR1 medium to ERK-independent naïve pluripotency for 96 hours in the 4i reprogramming medium6, which is a modified naïve human stem cell medium (NHSM)5. Our attempts to generate hPGCLCs starting with the same human iPSC clones but maintained in other commercially available human iPSC growth media before culture in the 4i reprogramming medium resulted in varying degrees of lower hPGCLC yields. Although whether longer-term adaptation in other media improves hPGCLC production or not remains be determined in future studies, this observation suggests that the exact state of the primed pluripotency of human iPSCs before the 4i reprogramming significantly impacts EB formation in the 4i medium and EB differentiation in hPGCLC medium.
Production of hPGCLCs from hiPSCs following our protocol is robust and highly reproducible, partly owing to the use of the microwell plates that enable efficient production of a large number of EBs (~8,000 EBs per batch) with a uniform size (3,000 hiPSCs per EB). The number of EBs readily produced in a single batch of experiment using our protocol may be far greater than methods using the regular U-bottom cell culture wells. Production of a large number of equally sized EBs uniformly studded with hPGCLCs may provide unique opportunities of high-throughput chemical screenings to identify small-molecular-weight activators or inhibitors affecting PGC specification or their biological characteristics such as epigenetic reprogramming. Such EBs may also be useful for toxicological assessments of large numbers of germline cell toxicants, including not only environmental pollutants but also clinically prescribed medications such as chemotherapeutic agents.
The critical factors of the robust and reproducible production of hPGCLCs using the presented protocol include (i) the use of healthy hiPSCs maintained in mTeSR1 on extracellular matrix protein, (ii) to inoculate exact number of cells as specified and strictly follow the timings of medium change and subculture, (iii) to select a good lot of human recombinant BMP4, and (iv) to minimize physical damages of EBs during the rocking culture. It is our experience that the best lot of BMP4 reagent worked at 100 ng/mL concentration whereas other lot of BMP4 reagents required 2X or greater doses. On the other hand, yield of hPGCLCs production using the best lot of BMP4 rather decreased at higher doses of BMP4 (e.g., 200 ng/mL). We recommend to test several different lots of recombinant human BMP4 reagents obtained from multiple vendors for their performance in supporting hPGCLC generation and to secure a large amount of the best lot.
A unique feature of our hPGCLC protocol is that hPGCLCs are localized on the outermost surface layer of EBs10 (Figure 8), whereas other protocols can generate hPGCLCs in the middle of cell aggregates6,8. Embryoid bodies tend to form multiple distinct layers such as surface, outer shell, inner shell, and core, and the central core regions are often necrotic due to limited supply of nutrients, oxygen, as well as pro-surviving growth factors provided form the culture medium by diffusion20. Localization of hPGCLCs on the surface of EBs without possible restrictions due to limited diffusion towards the center of EBs may be beneficial for direct, time- and dose-controlled exposure of hPGCLCs to drugs or toxic substances for pharmacological or toxicological studies.
Whereas mouse PGCLCs show robust genome-wide DNA demethylation involving the imprinting control regions at least partly7,19,20, the degree of global gDNA demethylation in hPGCLCs seems weaker than mouse PGCLCS or PGCs6,7. Transcriptomal profiles suggest that hPGCLCs may resemble an earlier stage of embryonic PGCs than mouse PGCLCs10. It has been reported that prolonged culture of EBs under the condition of hPGCLC production caused an increased degree of gDNA demethylation7; however, whether an extended period of culture of hPGCLCs in EBs or as isolated cells can achieve more advanced stages of germline differentiation needs to be determined by future studies.
Offenlegungen
The authors have nothing to disclose.
Acknowledgements
We acknowledge Shiomi Yawata and Chie Owa for technical assistance during initial studies. This study was supported by NIEHS/NIH grants R01 ES023316 and R21ES024861 to TS, and by Flight Attendant Medical Research Institute (FAMRI) grant to JHH.
Materials
| Primed pluripotency hiPSC culture | |||
| Matrigel | Corning | 354277 | Aliquot Matrigel at the volume indicated by the manufacturer (about 200 μL). Use cold tubes. Store at -80 °C |
| DMEM/F-12 | Thermo Fisher Scientific | 21041-025 | Store at 4 °C. |
| mTeSR1 | STEMCELL Technologies | 85850 | Reconstitute by adding 5X Supplement to Basal Medium. The reconstituted medium can be stored at 4 °C for up to 4 weeks without affecting cell culture performance. |
| Y27632 | Axon | 1683 | Dilute 5 mg Y27632 (MW 320.26) with 312.2 μL water to prepare 50 mM Y27632 stock solution. Sterilize by filtration (0.22 μm). Aliquot 20 μL/tube x 15 tubes and store at -20 °C. |
| CryoStor CS10 | STEMCELL Technologies | 07930 | Store at 4 °C. |
| Accutase | Innovative cell technologies | AT104-500 | Cell dissociation enzyme; aliquot 40 mL/tube x 12 tubes and store at -20 °C. |
| Name | Company | Catalog Number | Comments |
| 4i hiPSC culture | |||
| KnockOut DMEM | Thermo Fisher Scientific | A1286101 | |
| KnockOut Serum Replacement | Thermo Fisher Scientific | 10828028 | Aliquot 40 mL/tube x 12 tubes and store at -20 °C. |
| Penicillin-Streptomycin-Glutamine (100x) | Thermo Fisher Scientific | 10378016 | |
| MEM Non-Essential Amino Acids Solution (100x) | Thermo Fisher Scientific | 11140050 | |
| Insulin from bovine pancreas | Sigma-Aldrich | I1882-100MG | Add 0.1 mL glacial acetic acid to 10 mL water. Sterilize by filtration (0.22 um). Dilute 100 mg insulin lyophilized powder with cold 10 mL the acidified water to make 10 mg/mL stock solution. Aliquot 650 μL/tube x 15 tubes and store at -80 °C. |
| human LIF | PeproTech | 300-05 | Reconstitute 250 μg human LIF with 250 μL water. Add 750 μL of 0.1% bovine serum albumin in PBS(-) to make 250 μg/mL human LIF stock solution. Specific activity of this product is >10,000 units/μg. Aliquot 40 ul/tube x 25 tubes and store at -80 °C. |
| human FGF2 | R&D Systems | 4114-TC | Reconstitute 1 mg human FGF2 with 5 mL PBS(-) to make 200 μg/mL human FGF2 stock solution. Aliquot 100 μL/tube x 50 tubes and store at -80 °C. |
| human TGF-β1 | PeproTech | 100-21 | Reconstitite 50 μg with 50 μL of 10 mM Citric Acid (pH 3.0). Add 150 μL of 0.1% bovine serum albumin in PBS(-) to make 250 μg/mL human TGF-β1 stock solution. Aliquot 4 μL/tube x 50 tubes and store at -80 °C. |
| 4i basal medium | Mix the following reagents to make 4i basal medium. 500 mL of KnockOut DMEM 100 mL of KnockOut Serum Replacement 6 mL of Penicillin-Streptomycin-Glutamine (100x) 6 mL of MEM Non-Essential Amino Acids Solution (100x) 650 µL of 10 mg/mL Insulin from bovine pancreas 40 µL of 250 µg/mL human LIF 20 µl of 200 µg/ml human FGF2 4 µl of 250 µg/ml human TGF-β1 Aliquots 40 ml/tube and store at -80 °C. |
||
| CHIR99021 | Axon | 1386 | Reconstitute 25 mg CHIR99021 (MW 465.34) with 1791 μL DMSO to make 30 mM CHIR99021 stock solution. Aliquot 50 μL/tube x 35 tubes and store at -20 °C. |
| PD0325901 | Axon | 1408 | Reconstitute 5 mg PD0325901 (MW 482.19) with 1037 μL DMSO to make 10 mM PD0325901 stock solution. Aliquot 50 μL/tube x 20 tubes and store at -20 °C. |
| BIRB796 | Axon | 1358 | Reconstitute 10 mg BIRB796 (MW 527.66) with 948 μL DMSO to make 20 mM BIRB796 stock solution. Aliquot 50 μL/tube x 18 tubes and store at -20 °C. |
| SP600125 | Tocris | 1496 | Reconstitute 10 mg SP600125 (MW220.23) with 908 μL DMSO to make 50 mM SP600125 stock solution. Aliquot 50 μL/tube x 18 tubes and store at -20 °C. |
| AggreWell400 | STEMCELL Technologies | 27845 | Microwell plate |
| Pluronic F-127 | Sigma-Aldrich | P2443 | Reconstitute 5 g Pluronic F-127 in 100 mL water to make 5%(w/v) Pluronic F127 stock solution. Sterilize by filtration (0.22 um). Aliqout 50 mL/tube x 2 tubes and store at r.t. |
| Name | Company | Catalog Number | Comments |
| hPGCLC culture | |||
| Glasgow’s MEM | Thermo Fisher Scientific | 11710035 | |
| Sodium pyruvate (100 mM) | Thermo Fisher Scientific | 11360070 | |
| Penicillin-Streptomycin (100x) | Corning | 30-002-CI | |
| hPGCLC basal medium | Mix the following reagents to make hPGCLC basal medium. 500 mL of Glasgow’s MEM 75 mL of KnockOut Serum Replacement 6 mL of MEM Non-Essential Amino Acids Solution (100x) 6 mL of 100 mM Sodium pyruvate 6 mL of Penicillin-Streptomycin (x100) Aliquots 40 mL/tube and store at -20 °C. |
||
| 2-Mercaptoethanol | Sigma-Aldrich | M3148 | Dilute 350 μL 2-Mercaptoethanol (14.3 M) in 9.65 mL PBS(-) to make 500 mM 2-Mercaptoethanol stock solution . Store at 4 °C. |
| L-Ascorbic acid | Sigma-Aldrich | A4544 | Reconstitute 400 mg L-Ascorbic acid with 20 mL water to make 20 mg/mL L-Ascorbic acid stock solution. Sterilize by filtration (0.22 um). Aliquot 500 μL/tube x 40 tubes and store at -20 °C. |
| Recombinant human BMP4 | R&D Systems | 314-BP | Reconstitute 1 mg recombinant human BMP4 with 10 mL of 4mM HCl aq. to make 100 μg/mL recombinant human BMP4 stock solution. Aliquot 250 μL/tube x 40 tubes and store at -80 °C. |
| Human SCF | PeproTech | 300-07 | Reconstitute 250 μg human SCF with 500 μL of 0.1% bovine serum albumin in PBS(-) to make 500 μg/mL human SCF stock solution. Aliquot 35 μL/tube x 14 tubes and store at -80 °C. |
| Human EGF | PeproTech | AF-100-15 | Reconstitute 1 mg human EGF with 1 mL of 0.1% bovine serum albumin in PBS(-) to make 1 mg/mL human EGF stock solution. Aliqot 100 μL/tube x 10 tubes. Dilute 100 μL of 1 mg/ml human EGF stock solution with 300 μL of 0.1% bovine serum albumin in PBS(-) to make 250 μg/mL humanEGF stock solution. Aliquot 20 μL/tube x 20 tubes. Store at -80 °C. |
| Cell strainer | Corning | 352340 | Pore size = 40 μm. |
| 50 ml polypropylene conical tube | Corning | 352070 | |
| Low attachment plate | Corning | 3471 | |
| Vari-Mix Platform Rocker | Thermofisher | M79735Q | |
| Name | Company | Catalog Number | Comments |
| Immunohistochemical staining | |||
| BLOXALL Blocking Solution | VECTOR | SP-6000 | |
| Normal Horse serum ImmPRESS Reagent Anti-Goat IgG |
VECTOR | MP-7405 | |
| OCT-3/4 Antibody | Santa Cruz | SC-8629 | |
| ImmPACT DAB | VECTOR | SK-4105 | |
| Permount | Thermofisher | SP15-100 | Mounting medium |
| Name | Company | Catalog Number | Comments |
| Isolating hPGCLC | |||
| Embryoid Body Dissociation Kit | Miltenyi Biotec | 130-096-348 | |
| Anti-CD38 antibody conjugated to APC | abcam | ab134399 |
Referenzen
- Magnúsdóttir, E., Surani, M. A. How to make a primordial germ cell. Development. 141 (2), 245-252 (2014).
- Saitou, M., Yamaji, M. Primordial germ cells in mice. Cold Spring Harbor Perspectives in Biology. 4 (11), a008375 (2012).
- De Felici, M. Origin, Migration, and Proliferation of Human Primordial Germ Cells. Oogenesis. (Chapter 2), 19-37 (2012).
- Weinberger, L., Ayyash, M., Novershtern, N., Hanna, J. H. Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nature reviews. Molecular Cell Biology. 17, 155-169 (2016).
- Gafni, O., Weinberger, L., et al. Derivation of novel human ground state naive pluripotent stem cells. Nature. 504 (7479), 282-286 (2013).
- Irie, N., Weinberger, L., et al. SOX17 is a critical specifier of human primordial germ cell fate. Cell. 160 (1-2), 253-268 (2015).
- von Meyenn, F., Berrens, R. V., et al. Comparative Principles of DNA Methylation Reprogramming during Human and Mouse In vitro Primordial Germ Cell Specification. Developmental Cell. 39 (1), 104-115 (2016).
- Sasaki, K., Yokobayashi, S., et al. Robust In vitro Induction of Human Germ Cell Fate from Pluripotent Stem Cells. Cell Stem Cell. 17 (2), 178-194 (2015).
- Sugawa, F., Araúzo-Bravo, M. J., et al. Human primordial germ cell commitment in vitro associates with a unique PRDM14 expression profile. The EMBO Journal. 34 (8), 1009-1024 (2015).
- Mitsunaga, S., Odajima, J., et al. Relevance of iPSC-derived human PGC-like cells at the surface of embryoid bodies to prechemotaxis migrating PGCs. Proceedings of the National Academy of Sciences of the United States of America. 114 (46), E9913-E9922 (2017).
- Kojima, Y., Sasaki, K., et al. Evolutionarily Distinctive Transcriptional and Signaling Programs Drive Human Germ Cell Lineage Specification from Pluripotent Stem Cells. Cell Stem Cell. 21 (4), 517-532 (2017).
- Irie, N., Surani, M. A. Efficient Induction and Isolation of Human Primordial Germ Cell-Like Cells from Competent Human Pluripotent Stem Cells. Methods in Molecular Biology. 1463 (Chapter 16), 217-226 (2017).
- Magnúsdóttir, E., Dietmann, S., et al. A tripartite transcription factor network regulates primordial germ cell specification in mice. Nature Cell Biology. 15 (8), 905-915 (2013).
- Chen, D., Liu, W., et al. Germline competency of human embryonic stem cells depends on eomesodermin. Biology of Reproduction. 97 (6), 850-861 (2017).
- Saitou, M., Miyauchi, H. Gametogenesis from Pluripotent Stem Cells. Cell Stem Cell. 18 (6), 721-735 (2016).
- Hayashi, K., Ohta, H., Kurimoto, K., Aramaki, S., Saitou, M. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell. 146 (4), 519-532 (2011).
- Kobayashi, T., Zhang, H., et al. Principles of early human development and germ cell program from conserved model systems. Nature. 546 (7658), 416-420 (2017).
- Choi, J., Huebner, A. J., et al. Prolonged Mek1/2 suppression impairs the developmental potential of embryonic stem cells. Nature. 548 (7666), 219-223 (2017).
- Miyoshi, N., Stel, J. M., et al. Erasure of DNA methylation, genomic imprints, and epimutations in a primordial germ-cell model derived from mouse pluripotent stem cells. Proceedings of the National Academy of Sciences of the United States of America. 113 (34), 9545-9550 (2016).
- Shirane, K., Kurimoto, K., et al. Global Landscape and Regulatory Principles of DNA Methylation Reprogramming for Germ Cell Specification by Mouse Pluripotent Stem Cells. Developmental Cell. 39 (1), 87-103 (2016).
