For the quantification of DENV RNA by RT-qPCR analysis, a standard of the known copy number, which can be detected by the same primer set, is a prerequisite. In this protocol, the 462 nucleotide-long RNA containing the 3'UTR sequence of the DENV-2 NGC strain was in vitro transcribed from the T7 RNA promoter-fused DENV-2 3'UTR DNA template, which had been amplified by PCR and purified (Figure 2A and 2B). When a serial 10-fold dilution of the standard DENV RNA (from 5 x 109 to 5 x 102 copies in a 10 μL RT-qPCR reaction) was subjected to a direct RT-qPCR analysis using 3'UTR-specific primers and a fluorogenic probe9, a linear curve with a good correlation coefficient (R2 = 0.98726) was obtained (Figure 2C).
Next, this direct RT-qPCR assay was applied to quantify the DENV in the culture supernatant of virus-infected cells. DENV-2 (Singapore isolate EDEN2 329510), which had been propagated in C6/36 mosquito cells and titrated by plaque assay using BHK-21 cells11, was serially diluted (from 8 x 106 to 80 plaque formation units [PFU]/mL). DENV samples were then treated with an equal volume of processing buffer containing proteinase K to deproteinize virions and subjected to a direct RT-qPCR assay targeting the DENV 3'UTR RNA sequence. Again, a good correlation (R2 = 0.99981) between the DENV infectious titer and the Ct, a cycle number that is considered to be the point where the fluorescent signal rises with exponential growth above the background, was obtained (Figure 3A). When Ct values generated from a serial dilution of the DENV stock with known infectious titers were plotted on the standard curve made with in vitro transcribed 3'UTR RNA, all of the plots obtained from 8 x 106 to 80 PFU/mL (equal to 8 x 103 to 8 x 10-2 PFU in a 10 μL RT-qPCR reaction) were within the range of those subjected to standard RNA (5 x 109 to 5 x 103 copies in a 10 μL RT-qPCR reaction, Figure 3B), indicating that DENV samples with a wide range of infectious titers can be analyzed at the same time, using this direct RT-qPCR. In parallel experiments, the effect of processing buffer or proteinase K treatment on the detection of viral RNA by RT-qPCR analysis was investigated. Although treating a log dilution of the DENV sample (8 x 106 to 8 x 102 PFU/mL) with phosphate-buffered saline (PBS) alone prior to RT-qPCR (1:1 dilution of the virus sample with PBS and an incubation of 25 °C for 10 min and 75 °C for 5 min) elicited a regression curve (Figure 3C, black line), and the mean Ct values obtained by the PBS treatment were delayed 2.6–3.2 cycles when compared to the Ct obtained by processing buffer/proteinase K treatment (Figure 3C, dotted line). Additionally, the highest dilution of the virus (8 x 102 PFU/mL [8 x 10-1 PFU per 10 μL RT-qPCR reaction]) could not be detected with this PBS treatment (Figure 3C). In the absence of proteinase K during the processing buffer treatment (Figure 3C, gray line), a similar regression was generated; however, the delay in amplification (0.1–0.8 cycles) was still observed relative to the processing reaction containing proteinase K (Figure 3C, dotted line). These data indicate that, among the conditions tested, treatment of the DENV sample with processing buffer together with proteinase K improves the sensitivity of the RT-qPCR assay.
The direct RT-qPCR assay was further assessed for its applicability to validating antiviral agents against DENV. Mycophenolic acid (MPA), a nonnucleoside inhibitor of inosine monophosphate dehydrogenase that is used as an immunosuppressant in transplantation, has been reported to inhibit DENV in vitro12,13. Although the previous studies employed a conventional plaque assay or a flow cytometry assay to detect viral antigens to demonstrate the inhibitory effect of MPA on DENV infection12,13, in the present study, direct RT-qPCR was applied to evaluate the MPA's antiviral activity. HeLa cells, which had been seeded at a density of 5 x 104 cells/well in a 24-well plate 1 day prior to infection, were exposed to DENV-2 at an MOI of 1 for 1 h and, after washing, cultured with DMEM/10% FBS in the presence of 50–0.016 μg/mL MPA (or 0.1% dimethyl sulfoxide [DMSO]). The culture supernatant was collected 3 days after infection and subjected to the direct RT-qPCR assay using DENV 3'UTR specific primers and a fluorescent probe. Figure 4 shows the DENV RNA copies (per reaction) in the culture supernatants of infected cells treated with increasing concentrations of MPA, which were determined by an in vitro transcribed 3'UTR RNA standard. With a treatment of 50 μg/mL (156 μM) MPA, a reduction to 99.87 ± 0.02% of the DMSO-treated control culture was observed (Figure 4). Importantly, the IC50 (50% inhibitory concentration) value for MPA determined by the data shown in Figure 4 was 0.79 μM, which is similar to the values reported previously12,13. This result, therefore, indicates that this direct RT-qPCR assay is a useful and reliable method for assessing the inhibitory effect of antiviral agents on DENV infection.
Finally, applications of the direct RT-qPCR assay to other RNA viruses were tested. When a stock of yellow fever virus (YFV) 17D vaccine strain (Flaviviridae), which had been amplified in Vero cells and titrated on BHK-21 cells, was subjected to direct RT-qPCR using YFV-17D-specific primers and a fluorogenic probe14, as described in Table 1, a standard curve with good direct correlation between virus titers and Ct values could be generated with a 10-fold serial dilution of the virus stock (3.2 x 105 to 3.2 PFU/mL, Figure 5A). Likewise, direct RT-qPCR analysis of Chikungunya virus (CHIKV, Togaviridae) Ross strain stock, which had been amplified in Vero cells and titrated on BHK-21 cells, using CHIKV-specific primers and a fluorogenic probe15 (Table 1), gave a good regression between infectious titers (4.4 x 108 to 4.4 x 103 PFU/mL) and Ct values (Figure 5B). This was also the case for the detection of a serial dilution (8 x 102 to 8 x 10-2 PFU/mL, Figure 5C) of the measles virus (MeV, Paramyxoviridae) that had been propagated and titrated with Vero cells, using previously reported primers and a fluorogenic probe16 (Table 1). These data demonstrate the adaptability of the direct RT-qPCR assay for the quantitative detection of various RNA viruses.
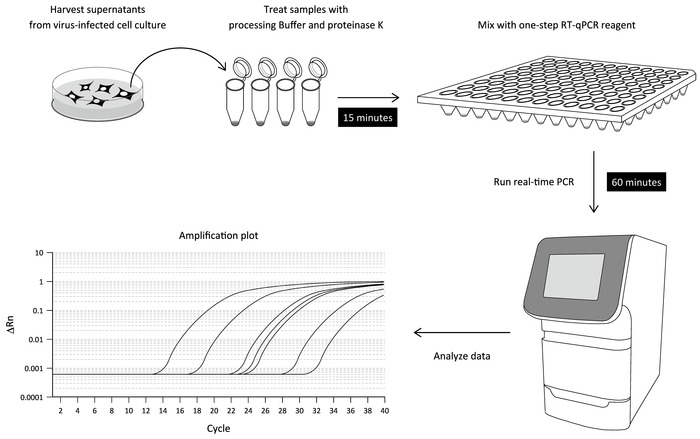
Figure 1: Workflow of the direct RT-qPCR assay. The culture supernatant of DENV-infected cells is treated with a processing buffer containing proteinase K to release viral RNA (sample-processing step). The processed sample is then mixed with a one-step RT-PCR reagent and subjected to the real-time PCR assay using DENV 3'UTR-specific primers and a fluorogenic probe. The levels of viral RNA detected in the respective samples can be determined by a serially diluted standard using in vitro transcribed DENV RNA. Please click here to view a larger version of this figure.
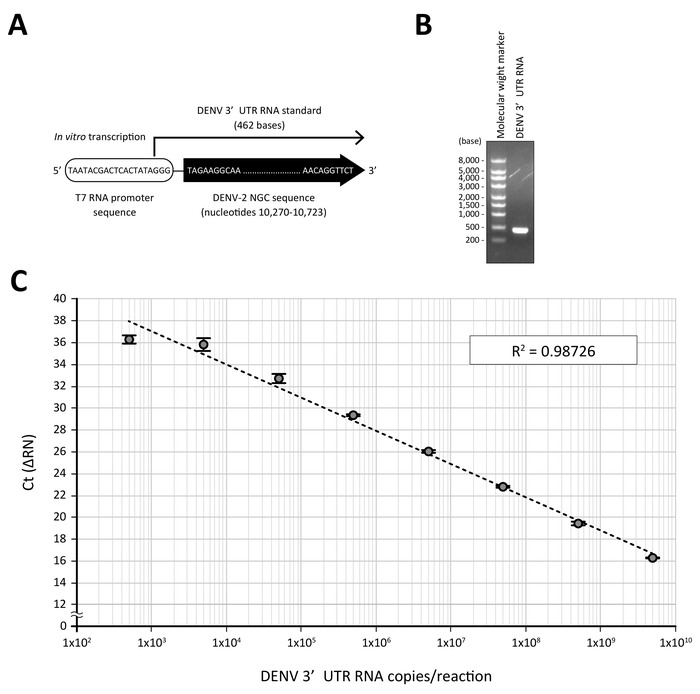
Figure 2: A standard curve generated by DENV 3'UTR RNA. (A) Standard RNA containing 3'UTR of DENV-2 NGC was transcribed in vitro from a T7 RNA promoter sequence-fused PCR fragment. (B) Purified DENV 3'UTR RNA (250 ng) was visualized on a 1% agarose gel (right lane). The left lane shows the molecular weight marker for RNA electrophoresis. (C) A log dilution of the DENV 3'UTR RNA standard was treated with the processing buffer containing proteinase K and subjected to a real-time PCR analysis using a one-step RT-qPCR reagent and DENV 3'UTR-specific primers and a fluorogenic probe set. The mean Ct values (n = 3) obtained in respective dilutions were plotted against the estimated quantity of 3'UTR RNA (5 x 109 to 5 x 102 RNA copies in a 10 μL RT-qPCR reaction). R2 is the correlation coefficient. Please click here to view a larger version of this figure.
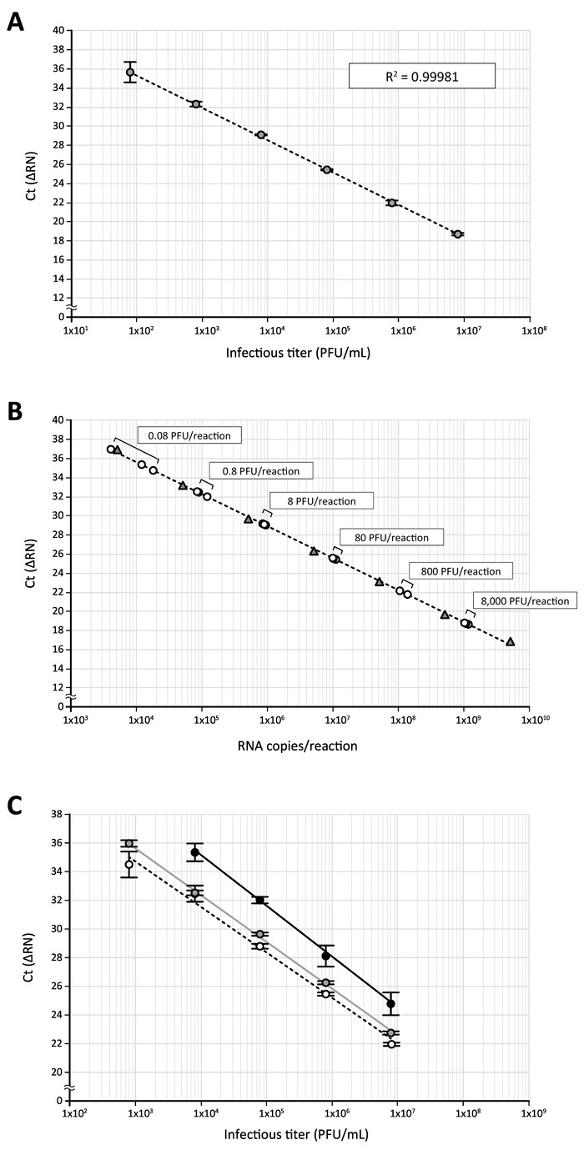
Figure 3: The quantification of DENV RNA in virus stock by direct RT-qPCR. (A) The high-titer DENV-2 stock produced in C6/36 cells was serially diluted 10-fold (8 x 106 to 8 x 101 PFU/mL) with DMEM/10% FBS containing an RNase inhibitor and subjected to the direct RT-qPCR assay using DENV 3'UTR-specific primers/probe. (B) Ct values obtained by RT-qPCR of log-diluted DENV stock (circles) were plotted on a standard curve of the 3'UTR RNA (triangles) transcribed in vitro. The use of 8 x 106 PFU/mL stock in a direct RT-qPCR assay corresponds to 8 x 103 PFU of virus in a 10 μL RT-qPCR reaction. (C) This panel shows the effect of processing buffer and proteinase K treatments on the detection of viral RNA. DENV diluted stock (8 x 106 to 8 x 102 PFU/mL) was incubated with an equal volume of processing buffer containing proteinase K (white circles), processing buffer alone (gray circles), or PBS (black circles) and then subjected to the direct RT-qPCR assay. Please click here to view a larger version of this figure.
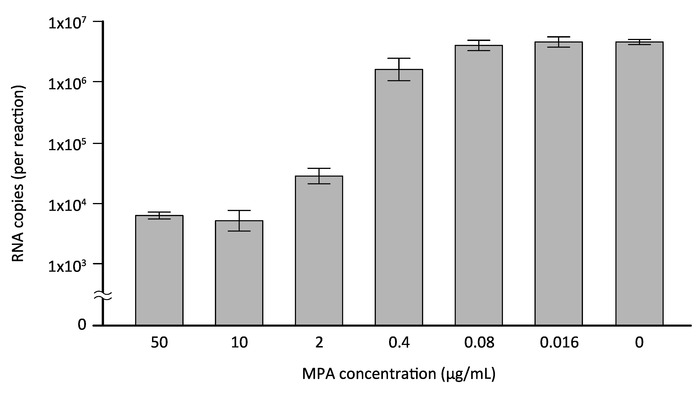
Figure 4: Evaluation of the inhibitory effect of MPA by direct RT-qPCR. HeLa cells were infected with DENV-2 at a MOI of 1 and cultured in the presence of increasing concentrations of MPA, a previously reported inhibitor of DENV (or 0.1% DMSO). Three days after infection, culture supernatants of the infected cells were collected and subjected to the direct RT-qPCR assay using DENV 3'UTR-specific primers/probe. The copy number of viral RNA was determined by DENV 3'UTR RNA standard transcribed in vitro. Please click here to view a larger version of this figure.
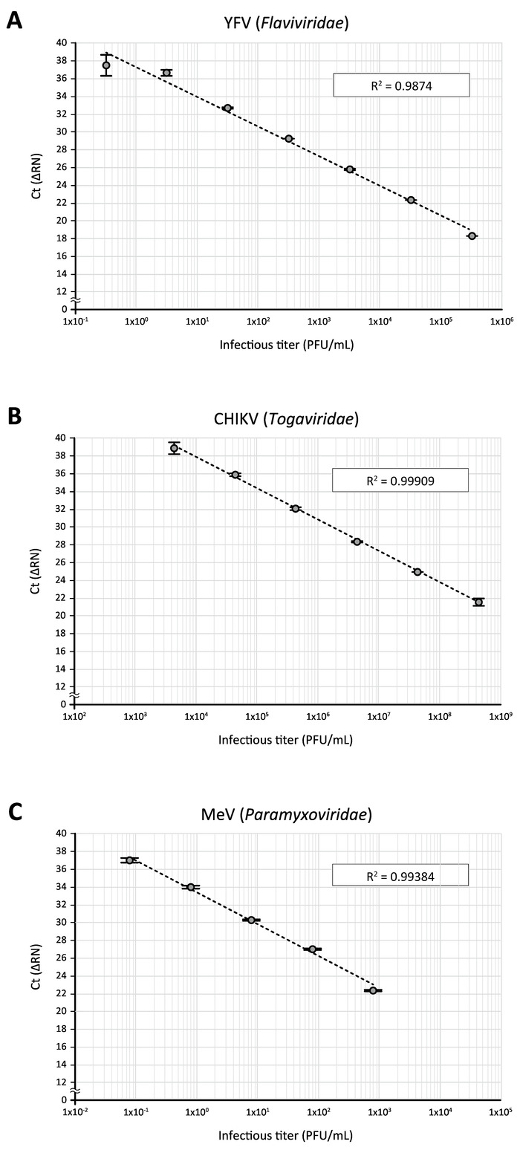
Figure 5: The application of direct RT-qPCR for the detection of other RNA viruses. Serially diluted virus stocks of (A) YFV, (B) CHIKV, and (C) MeV were subjected to the direct RT-qPCR assay using PCR primers and fluorogenic probes specific to their respective viral RNA sequences. Please click here to view a larger version of this figure.
| Purpose | Name | Sequence (5' – 3') | Note |
| T7 promoter-fused DENV-2 3'UTR cDNA amplification | T7-DENV 3’UTR Fwd | TAA TAC GAC TCA CTA TAG GGc gaa tTA GAA GGC AAA ACT AAC ATG AAA | Italic,T7 promoter; lower-case, spacer; bold, DENV-2 NGC nucleotides 10,270-10,292 |
| DENV 3’UTR Rvs | AGA ACC TGT TGA TTC AAC AGC | DENV-2 NGC nucleotides 10,723-10,703 | |
| RT-qPCR analysis of DENV | DENV-2 3'UTR F | AAG GAC TAG AGG TTA GAG GAG ACC C | Reference 9 |
| DENV-2 3'UTR R | GGC GTT CTG TGC CTG GAA TGA TG | ||
| Probe 2 Den-2-4 | 6FAM-AAC AGC ATA TTG ACG CTG GGA AAG ACC-TAMRA | ||
| RT-qPCR analysis of YFV | YFV NS5 F | GAA CAG TGA TCA GGA ACC CTC TCT | Reference 14 |
| YFV NS5 R | GGA TGT TTG GTT CAC AGT AAA TGT G | ||
| YFV NS5 Probe | 6FAM-CTA CGT GTC TGG AGC CCG CAG CAA T-TAMRA | ||
| RT-qPCR analysis of CHIKV | CHIK E1 F | TCG ACG CGC CCT CTT TAA | Reference 15 |
| CHIK E1 R | ATC GAA TGC ACC GCA CAC T | ||
| CHIK E1 P | 6FAM-ACC AGC CTG CAC CCA TTC CTC AGA C-TAMRA | ||
| RT-qPCR analysis of MeV | MeV N F | TGG CAT CTG AAC TCG GTA TCA C | Reference 16 |
| MeV N R | TGT CCT CAG TAG TAT GCA TTG CAA | ||
| MeV N P | 6FAM-CCG AGG ATG CAA GGC TTG TTT CAG A-TAMRA |
Table 1: Oligonucleotide primer and fluorogenic probe sequences used in this study.
| Components | Stock concentration | Volume (μL) | Final concentration |
| PrimeSTAR Max Premix | 2x | 25 | 1x |
| T7-DENV 3’UTR Fwd | 1 μM | 10 | 200 nM |
| DENV 3’UTR Rvs | 1 μM | 10 | 200 nM |
| pEU/DENV 3'UTR | 1 ng/μL | 1 | 20 pg/μL |
| Nuclease-free H2O | 4 | ||
| Total | 50 |
Table 2: Components of a PCR mix for the preparation of DENV 3'UTR template DNA.
| Components | Stock concentration | Volume (μL) | Final concentration |
| T7 sequence-fused DENV 3’UTR DNA template | (variable) | x | 100 ng per reaction |
| ATP solution | 75 mM | 2 | 7.5 mM |
| CTP solution | 75 mM | 2 | 7.5 mM |
| GTP solution | 75 mM | 2 | 7.5 mM |
| UTP solution | 75 mM | 2 | 7.5 mM |
| Reaction Buffer | 10x | 2 | 1x |
| T7 Enzyme Mix | 2 | ||
| Recombinant RNase Inhibitor | 40 units/μL | 1 | 2 units/μL |
| Nuclease-free H2O | 7 – x | ||
| Total | 20 |
Table 3: Components of an IVT mix for synthesizing the DENV 3'UTR RNA standard.
| Components | Stock concentration | Volume (μL) | Final concentration in an RT-qPCR reaction |
| iTaq universal probes reaction mix | 2x | 5.00 | 1x |
| iScript advanced reverse transcriptase | 40x | 0.25 | 100 ng per reaction |
| Primer/probe mix | DENV-2 3'UTR F: 3 μM | 1.00 | 300 nM |
| DENV-2 3'UTR R: 3 μM | 300 nM | ||
| Probe 2 Den-2-4: 2 μM | 200 nM | ||
| Nuclease-free H2O | 1.75 | ||
| Subtotal | 8.00 |
Table 4: Components of a master mix for real-time RT-qPCR analysis of DENV RNA. Note that 2 μL of the processed DENV sample (or standard RNA) should be added to the 8-μL master mix (a total of 10 μL per reaction).
