Preparation and Gene Modification of Nonhuman Primate Hematopoietic Stem and Progenitor Cells
Summary
The goal of this protocol is to isolate nonhuman primate CD34+ cells from primed bone marrow, to gene-modify these cells with lentiviral vectors, and to prepare a product for infusion into the autologous host. The total protocol length is approximately 48 h.
Abstract
Hematopoietic stem and progenitor cell (HSPC) transplantation has been a cornerstone therapy for leukemia and other cancers for nearly half a century, underlies the only known cure of human immunodeficiency virus (HIV-1) infection, and shows immense promise in the treatment of genetic diseases such as beta thalassemia. Our group has developed a protocol to model HSPC gene therapy in nonhuman primates (NHPs), allowing scientists to optimize many of the same reagents and techniques that are applied in the clinic. Here, we describe methods for purifying CD34+ HSPCs and long-term persisting hematopoietic stem cell (HSC) subsets from primed bone marrow (BM). Identical techniques can be employed for the purification of other HSPC sources (e.g., mobilized peripheral blood stem cells [PBSCs]). Outlined is a 2 day protocol in which cells are purified, cultured, modified with lentivirus (LV), and prepared for infusion back into the autologous host. Key readouts of success include the purity of the CD34+ HSPC population, the ability of purified HSPCs to form morphologically distinct colonies in semisolid media, and, most importantly, gene modification efficiency. The key advantage to HSPC gene therapy is the ability to provide a source of long-lived cells that give rise to all hematopoietic cell types. As such, these methods have been used to model therapies for cancer, genetic diseases, and infectious diseases. In each case, therapeutic efficacy is established by enhancing the function of distinct HSPC progeny, including red blood cells, T cells, B cells, and/or myeloid subsets. The methods to isolate, modify, and prepare HSPC products are directly applicable and translatable to multiple diseases in human patients.
Introduction
Stem cell gene therapy is a powerful means to address a wide range of human pathologies. HSPC gene therapy is a particularly attractive approach, due to i) the relative ease of collecting these cells from patients, ii) the wealth of knowledge that is available regarding cell surface phenotypes and ex vivo culture parameters, and, as the field expands, because iii) it presents scientists with an ever-increasing toolbox of gene modification strategies tailored to various diseases of interest. We are actively investigating HSPC gene therapy approaches from multiple angles, including the basic science of HSPC biology, the engraftment of gene-modified HSPCs in preclinical in vivo models, and the application to relevant patient populations. We and others have characterized the cell surface phenotype of functionally distinct HSPC subsets1,2,3, the mobilization and conditioning regimens that maximize HSPC yield and engraftment while minimizing toxicity4,5, and the gene modification and gene-editing strategies that have been tailored to a wide range of malignant, genetic, and infectious diseases6,7,8,9,10. The function and engraftment of gene-modified HSPCs can be evaluated in a number of small- and large-animal models, including mice, dogs, and NHPs. In particular, NHP models are advantageous because many reagents, for example, antibodies specific for HSPC cell surface proteins like CD34 and CD90, can be used interchangeably in human and NHP cells. Furthermore, in contrast to mice, large animals such as NHPs allow a closer approximation of the scale of gene modification necessary for clinical efficacy. Finally, NHPs are the gold standard for the modeling of human pathologies such as HIV-1 infection11 and are an emerging model system for candidate anticancer and anti-HIV immunotherapies12,13.
The purpose of this protocol is to outline methods for purifying, genetically modifying, and preparing NHP HSPC infusion products. Although outside the scope of this protocol, we have previously shown that these products engraft in autologous NHP hosts, give rise to all hematopoietic lineages, and provide therapeutic efficacy in a broad range of disease models1. We have also characterized the clonality of engrafting HSPCs and built a platform to track the kinetics, trafficking, and phenotype of individual HSPCs and their progeny, following autologous transplantation1,14. The methods presented here have been developed with the following goals: i) to isolate highly pure HSPCs and long-term engrafting HSC subsets, ii) to maintain primitive HSCs during ex vivo culture, and iii) to efficiently gene-modify either bulk HSPCs or long-term engrafting HSC subsets. We employ magnetic-assisted cell-sorting (MACS), as well as fluorescence-activated cell sorting (FACS), to isolate phenotypically/functionally distinct HSPC populations, consistent with the methods of many groups2,15,16. The maintenance of primitive HSCs in culture (i.e., minimizing the differentiation of these cells into committed progenitors that give rise to fully differentiated lymphoid and myeloid subsets) is an essential facet of the protocol described here. Although we have previously characterized approaches to expand HSPCs while retaining a primitive phenotype17,18, here, we describe a protocol that focuses on maintaining HSCs via a minimal (48 h) and defined ex vivo culture.
The efficient modification of HSPCs and HSC subsets is a central goal of this protocol. Among several approaches we have reported, two are by far the most investigated in clinical trials: LV-mediated gene modification and nuclease-mediated gene editing1,6,19. Gene-editing strategies use one of a number of nuclease platforms to specifically modify a targeted gene of interest, for example, C-C chemokine receptor type 5 (CCR5) for the treatment of HIV infection7,19 or Bcl11A for the treatment of hemoglobinopathies6. Here, we focus on LV-mediated gene modification, in which transgenic cargoes integrate semirandomly into the genome1,8,20. A key advantage of LV approaches is the ability to deliver large amounts of genetic material (up to 8 or 9 kilobases). Although gene-editing strategies are being developed to target a transgene of interest to integrate only at a specified locus by homologous donor recombination (HDR), these methods require further development in vitro and in small animal models. In contrast, LV vectors have been used extensively in NHPs and in patients21,22. Importantly, the protocol described here, which uses primed BM as a starting HSPC source, can be easily and broadly adapted, for example, to isolate PBSCs. As described above, we take advantage of the high degree of genetic similarity between NHPs and humans to use reagents that are applicable to both species. Finally, this approach has been adapted to modify other hematopoietic subsets, namely T cells12,23,24; the advent of efficacious T-cell immunotherapy approaches has relied heavily on the same LV platform utilized in this protocol. These methods are appropriate for any researcher interested in either HSPC biology or LV-mediated gene modification. For example, the HSPC purification protocol presented here could be used to characterize novel HSC-enriched subsets, as described previously1,15,25. Likewise, the LV transduction methods presented here could similarly be applied and further developed for numerous other cell types and experimental questions, both in in vitro and in vivo models.
In summary, we present methods to isolate and genetically modify NHP HSPCs. These methods can be easily adapted for other species and other sources of HSPCs. This thoroughly vetted protocol shows great promise in the modeling of efficacious therapies for numerous human diseases.
Protocol
Autologous NHP transplants, priming (mobilization), the collection of cells, and gene modification are conducted consistent with previously published protocols26. All experimental procedures are reviewed and approved by the Institutional Animal Care and Use Committee of the Fred Hutchinson Cancer Research Center and the University of Washington (Protocol #3235-01). All studies are carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (“The Guide”); animals were randomly assigned to the studies.
1. Enrichment of CD34+ HSPCs and Overnight Culture (Day -1)
- Harvest BM and condition it.
- Mobilize NHPs with granulocyte colony-stimulating factor (GCSF) for 4 days as described previously26.
- Sedate the animals with 100 mg/kg of ketamine and 0.03 mL/kg of dexmedetomidine (0.5 mg/mL stock). Administer analgesics (e.g., Buprenorphine SR) at the time of the draw.
- Using clippers, shave the animal’s hair from the proximal end of the humerus and/or the femur bone(s) and scrub the skin with iodine-based scrub or a similar antiseptic solution, alternate with alcohol, and repeat 3x. As an additional anesthetic, infuse the periosteum with bupivacaine (2 mg per site, 5 mg/mL stock).
- Position the bone marrow aspiration needle (16 G) over the periosteal entry site (medullary cavity) and pierce the skin, penetrating the medullary cavity using a rotating motion. Remove the stylet of the aspiration needle and reserve it in a sterile field for further use.
NOTE: Choose the periosteal entry site to the medullary cavity in an area of the bone that is not covered by muscles and where the shape/landscape of the bone is either visible or can be easily felt through the skin (commonly toward the proximal or distal end of the bone). - To the BM needle, attach a prefilled syringe containing a mixture of anticoagulant citrate dextrose solution (ACD-A) and heparin (20 USP/mL, 1 mL per 9 mL of marrow to be collected).
- Draw the plunger back while gently rocking the limb and rotating it, to agitate the syringe to mix aspirate and anticoagulant. Rotate the needle and reposition it throughout the aspiration and withdrawal to access a greater percentage of cells within the marrow space.
- Once the sample is collected, remove the needle and apply pressure to the aspiration site until the bleeding stops.
NOTE: Depending on the volume needed, bone marrow from one to four limbs (two humeri, two femurs) may be drawn during a single collection. No more than 20 mL should be collected from each limb, and the total volume collected should constitute no more than 10% of the animal’s weight (10 mL/kg). - If receiving aspirates from multiple limbs, combine and record the volume.
- Let the NHP recover postanesthesia.
- Reverse the effects of dexmedetomidine with 0.03 mL/kg atipamezole (5 mg/mL stock) after the procedure. Provide analgesics (e.g., Buprenorphine SR) as prescribed by clinical veterinary staff for at least 48 h after the procedure or longer, at the discretion of the clinical veterinarian, based on clinical signs.
- Return the animal to its home cage after the procedure and monitor it every 15 – 20 min, until the animal is able to sit up on its own. Monitor the animal daily by vet staff until it is determined that the aspiration site(s) are healed. In addition, monitor for any signs of inflammation, infection, or pain.
- In parallel to cell processing, condition the same NHP with myeloablative total body irradiation (TBI): 1,020 cGy at a rate of 7 cGy/min. Administer irradiation in fractionated doses over the 2 days before cell infusion.
- Hemolyze the BM.
- Divide the BM into 50 mL conical tubes (10 – 12 mL per tube) and add hemolytic buffer to bring the volume to 50 mL (Table 1). Incubate the cells at room temperature (RT) until they are lysed but no longer than for 7 min. Centrifuge at 800 x g for 5 min and aspirate the supernatant from the pellet(s), leaving 2 – 5 mL of volume/tube. Resuspend the pellet in the residual volume.
NOTE: Successfully lysed samples change color, from a dark red to a transparent light red. - Add 10 – 15 mL of hemolytic buffer/pellet. Remove any blood clots using 70 µm cell strainers. Add hemolytic buffer up to 50 mL, incubate for 5 min at RT, and spin at 800 x g for 5 min.
- Aspirate the supernatant and resuspend the cells in 10 mL of MACS buffer (Table 1). Filter again through a 70 µm cell strainer. Rinse the tube and filter it with 40 mL of MACS buffer up to 50 mL.
- Divide the BM into 50 mL conical tubes (10 – 12 mL per tube) and add hemolytic buffer to bring the volume to 50 mL (Table 1). Incubate the cells at room temperature (RT) until they are lysed but no longer than for 7 min. Centrifuge at 800 x g for 5 min and aspirate the supernatant from the pellet(s), leaving 2 – 5 mL of volume/tube. Resuspend the pellet in the residual volume.
- Enrich the CD34+ cells from the hemolyzed white blood cells (WBCs) using standard protocols.
- Spin the cells at 800 x g for 15 min. Check the tubes to make sure no cell swirls are evident in the top/middle. If cells are present above the pellet, spin again at a higher speed (up to 1,000 x g maximum) and for up to 10 min.
- Aspirate the supernatant and resuspend it in 10 mL or less of MACS buffer, noting the exact volume. Count the cells at a 1:100 or 1:1,000 dilution using a hemocytometer or an automated cell counter.
NOTE: This total WBC count is the pre-enrichment count. - Add MACS buffer to bring the cell concentration to 1 x 108 cells/mL. If necessary, first repeat steps 1.3.1 – 1.3.2 (e.g., due to a low pre-enrichment count). Reserve 5 x 106 cells in MACS buffer for HSC subset staining (pre-enrichment sample).
- Add unconjugated anti-CD34 antibody (clone 12.8, Table of Materials)27 to the remaining pre-enrichment cells to a final concentration of 40 µg/mL (1 x 108 cells/mL). Incubate the cell suspension at 4 °C on a tube rotator (see the Table of Materials) for 25 – 30 min.
- Use MACS buffer to bring the volume to 50 mL and spin the cells at 800 x g for 5 min. Aspirate the supernatant, resuspend the cells in 50 mL of MACS buffer, and spin again at 800 x g for 5 min.
- Degas 100 mL of MACS buffer with two filter tubes, wrapping the air inlet with wax paper, for 25 – 30 min or till ready to use.
- Calculate the necessary volume of magnetic beads: 1 mL of beads/109 cells. Aspirate the supernatant and resuspend the cells to 108 cells/mL, after the beads are added. For example: 3 x 109 cells + 3 mL of beads + 27 mL of MACS buffer to 30 mL of total volume.
- Incubate the cell suspension at 4 °C on a tube rotator for 25 – 30 min.
- During acquire magnets for the columns; plan to use 0.6 x 109 cells per column. Place the columns on the magnet and position a 50 mL conical tube below each column to collect the flow through. Prepare a 15 mL conical tube and the plunger for each column for elution (step
- When the incubation in step 1.3.8 is complete, add the MACS buffer to 50 mL and spin at 800 x g for 5 min. Aspirate the supernatant and resuspend the cells in degassed MACS buffer (2 mL per column). Pipette gently to avoid introducing bubbles.
- Without allowing the column to run dry, add 1 mL of degassed MACS buffer to each column, followed by 2 mL of cell suspension. Rinse the filter and original tube with degassed MACS buffer and divide the solution equally between columns (6 mL/column); then, add 7 mL of degassed MACS buffer for the final wash.
- Gently pull the column away from the magnet (push the top back) and collect the last drip into the flow-through tube. Add 5 mL of degassed MACS buffer to each column and apply the plunger to elute the cells into the sterile conical tube from step 1.3.9. Do not collect any liquid after bubbles appear.
- Count the cells in both the enriched and the flow-through (FT) collection tubes at a dilution ratio of 1:10. Keep the cells on ice. Aliquot 1 x 106 cells from the CD34-enriched fraction and 5 x 106 cells from the FT fractions for analysis by flow cytometry. Store the FT fraction at 4 °C until the quality control (step 2) is concluded.
NOTE: The number of CD34+ cells required for successful multilineage engraftment in the NHP is dependent on the frequency of the HSC-enriched CD34+CD90+CD45RA– subset. Based on an average frequency of 3% – 5% and the minimum requirement of 122,000 CD34+CD90+CD45RA– cells per kilogram of body weight1, it is recommended to proceed with a total of at least 2.5 x 106 and up to 10 x 106 CD34+ cells per kilogram of body weight. - Add MACS buffer to the CD34+ enriched fraction to a 50 mL in total, spin at 800 x g for 5 min, aspirate the supernatant, and resuspend the cells to 106 cells/mL in HSPC media (Table 1).
- Plate the cells at a density of 106 cells/mL in vented, tissue-culture (TC)-treated T-75 flasks, 10 – 20 mL per flask, and incubate them overnight in a 37 °C, 5% CO2 incubator. Rest the flasks lengthwise (not standing up).
NOTE: If desired, additional CD34+ cells or CD34– FT can be cryopreserved in 90% heat-inactivated fetal bovine serum (FBS) + 10% dimethyl sulfoxide (DMSO) at a concentration of 1 x 106 to 5 x 106 cells/mL, then slow-cooled at -80 °C (e.g., using freezing containers). The average recovery yield of cryopreserved cells is dependent on the CD34+ purity and commonly ranges from 50% to 80% with a viability of >80%.
- Plate the cells at a density of 106 cells/mL in vented, tissue-culture (TC)-treated T-75 flasks, 10 – 20 mL per flask, and incubate them overnight in a 37 °C, 5% CO2 incubator. Rest the flasks lengthwise (not standing up).
2. Quality Control of CD34-enriched Cells (Day -1)
- Prepare samples for flow cytometry.
- Resuspend samples reserved from each isolation stage mentioned above (pre-enrichment, FT, and CD34-enriched fractions) in FACS buffer at a concentration of 10 x 106 cells/mL and transfer 100 µL of each cell suspension (Table 1) to a FACS tube.
NOTE: Control samples should include unstained cells from each isolation stage (e.g., 5 x 105 cells) for the adjustment of forward scatter (FSC), side scatter (SSC), and autofluorescence in each fluorescence channel. In addition, single-stained compensation beads for each fluorochrome-conjugated antibody will be required to adjust for the compensation in between adjacent channels (Table 2 and Table 3). - Add desired antibodies (e.g., anti-CD45, anti-CD34, anti-CD45RA, and anti-CD90), according to the manufacturer’s instructions, for one test (1 x 106 cells in 100 µL) (Table 3). Incubate for 20 min at 4 °C. Add 3 mL of FACS buffer and spin at 800 x g for 5 min, aspirate the supernatant, resuspend in 100 µL of FACS buffer, and analyze on an appropriate flow cytometer (step 2.2).
- Resuspend samples reserved from each isolation stage mentioned above (pre-enrichment, FT, and CD34-enriched fractions) in FACS buffer at a concentration of 10 x 106 cells/mL and transfer 100 µL of each cell suspension (Table 1) to a FACS tube.
- Prepare the flow cytometer/cell sorter for analysis.
NOTE: It is recommended to perform the analysis on the same machine that will be used for cell sorting in step 2.3.- Generate a protocol including FSC/SSC, CD45/CD34, and CD45RA/CD90 (three flow plots). Right-click on a flow plot and add the Population hierarchy layout.
- Left-click on the FSC/SSC plot, select the polygon gate feature, and draw a gate to exclude debris and dead cells. Highlight the new gate and rename it to Scatter in the Inspector window.
NOTE: This is automatically termed as P1. - Right-click in the center of the CD45/CD34 flow plot, navigate to Show populations, and gate the plot on scatter. Draw a rectangular gate around the CD45intCD34+ cells to exclude non-CD34 cells and nonhematopoietic cells, as well as residual platelets and erythrocytes (CD45–). Rename the gate to CD34+.
- Gate the CD45RA/CD90 flow plot on the CD34+ population. Draw three independent subgates around CD90+CD45RA– cells (enriched for HSCs), CD90–CD45RA– cells (enriched for multipotent and erythro-myeloid progenitors [MPPs/EMPs]), and CD90–CD45RA+ cells (enriched for lympho-myeloid progenitors [LMPs]). Rename the gates to HSC, MPP-EMP, and LMP, respectively.
- When complete, ensure that the Population hierarchy contains six hierarchically organized entries in the following order: 1) All Events; 2) Scatter; 3) CD34+; 4) HSC; 5) MPP-EMP; 6) LMP. Ensure that the flow plots and the shape of the gate look as illustrated in Figure 4.
- Run unstained pre-enrichment WBCs to adjust the voltage for FSC, SSC, and all fluorescence channels (Table 2, sample 5). Run single-stained compensation beads to adjust the compensation in between adjacent channels (Table 2, samples 1 – 4). Run all remaining unstained and stained samples with the adjusted and compensated protocol, to document CD34 enrichment efficiency (Table 2, samples 6 – 9).
NOTE: Keep the final sample (Table 2, Sample 10) for the simultaneous flow analysis and cell sorting in step 2.4.
- Configure the cell sorter for the sort-purification of CD34 subsets into colony-forming cell (CFC) assays.
NOTE: If a different machine has been used for analysis (step 2.2), set up the protocol on the cell sorter as previously described in steps 2.2.1 – 2.2.5.- Adjust the angle/voltage for the left- and right-side streams in the cell sorter software to deposit cells into 15 mL tubes containing CFC medium. Configure the angle of the side stream to prevent misalignment. Step-wise, fine-tune the angle of the deflected side stream until the sorted cells hit the CFC medium and not the side of the tube because very few cells are sorted.
- In the Browser, open the Global Worksheets folder, and create a new sort-layout using the button in the top menu of the Browser window. Change the entry in the Collection Device drop-down menu to 2 tubes, the entry in the Precision drop-down menu to 4-way purity or Single cell and set the Target Events to 800 – 1,200 cells. Left-click on the sort-location field (Left or Right), select the Entry add in the menu, and choose the population to sort from the menu.
- Sort the cells for CFC assays.
NOTE: Sorting for the CFC assays is only performed with cells from the CD34-enriched product (Table 2, sample 10).- Load sample 10, record 2,000 – 3,000 events, and fine-adjust the sort gates to fit the signal strength and target populations.
- When the setup is complete, acquire cells (Table 2, sample 10). Acquire data as described in step 2.2.3, adjust the flow rate to 500 – 1,000 cells/s, and sort 800 – 1,200 cells from i) the Scatter gate, ii) the CD34+ gate, iii) the HSC gate, iv) the MPP-EMP gate, and v) the LMP gate into separate tubes containing 3.6 mL of CFC media.
NOTE: Sorting for CFC assays is only performed with cells from the CD34-enriched product (Table 3, sample 10). Cells from the Scatter and CD34+ gates must be sorted individually, whereas HSCs, MPP-EMPs, and LMPs can be sorted simultaneously into the left and right tube holder position. - Vortex and dispense 1 mL of cell suspension to each of the 3 x 3.5 cm sterile, nontissue, culture-treated Petri dishes. Incubate the cells at 37 °C for 10 – 14 days in a secondary container (e.g., a 15 cm Petri dish).
- On days 10 – 11 postplating, count individual colonies from all three plates per condition, based on colony morphology.
3. Gene Modification of CD34+ HSPCs and Overnight Recovery (Day 0)
- Dilute 2.5 mL of 1 µg/µL CH-296 solution to 50 µg/mL in 50 mL of Hank’s balanced salt solution (HBSS, see the Table of Materials).
- Prepare flasks for the LV transduction.
- Determine the approximate number of nontissue-culture (non-TC)-treated flasks needed (usually from the cell counts determined in step 1.4). Plan to plate 1 x 107 cells in 10 mL of media per T-75 flask (1 x 108 total cells would require 10 flasks) and include one non-TC-treated 12-well plate (mock transduction condition). Add sterile HBSS and 50 µg/mL CH-296 stock from step 3.1 to each flask/plate, at a concentration of 2 µg/cm2. For T-75 flasks, use 3 mL of 50 µg/mL CH-296 + 7 mL of HBSS; for the 12-well plate, use 160 µL of 50 µg/mL CH-296 + 340 µL of HBSS per well.
- Allow the dishes to sit undisturbed at RT on a clean bench or in the hood for 2 h. Aspirate the CH-296 and replace it with a similar volume of sterile HBSS + 2% bovine serum albumin (BSA). Incubate at RT for 30 min, aspirate the HBSS/BSA, and wash the dishes with a similar volume of sterile HBSS containing 2.5% 1 M HEPES, pH 7.0. Aspirate immediately prior to plating the cells (step 3.4).
NOTE: Following step 3.2.2, do not allow the plates/flasks to dry out. Plates containing sterile HBSS + 2.5% 1 M HEPES are held at 4 °C overnight (i.e., prepare on day -1).
- Harvest and transduce the CD34+ cells from day -1.
- Using a 10 mL pipette, rinse loosely adherent cells by the repeated, gentle rinsing of each culture flask with sterile HBSS. Make sure that all cells will detach (if necessary, tap/slap the flask to loosen the cells).
- Centrifuge the cells at 800 x g for 5 min, aspirate the supernatant, and resuspend the cells in transduction media to a concentration of 1 x 106 cells/mL. Once all cells have been collected, determine the cell count, using a hemocytometer or automated cell counter.
- Add 1 mL of cell suspension (1 x 106 cells) to one well of the CH-296-coated 12-well plate (for the mock-transduced control sample). Divide the remainder of the cells among the T-75 flasks (step 3.2.2), adding approximately 10 mL (1 x 107 cells) per flask. Allow the cells to adhere to the CH-296 coating by incubating at 37 °C, 5% CO2 for 30 min, with the caps vented.
- Thaw virus-conditioned media (VCM, Table of Materials) and determine the titer in infectious units per milliliter (IU/mL)28,29,30,31,32. Add the appropriate volume of VCM to each T-75 flask from step 3.3.3 and incubate the cells at 37 °C, 5% CO2.
NOTE: If performing a single transduction, incubate overnight; if performing a double transduction, repeat this stepapproximately 6 – 8 h after the first transduction without a wash/recovery phase and, then, incubate overnight. For example, for one flask (1 x 107 cells) with a desired multiplicity of infection (MOI) of 10, add 1 mL of virus titered at 1 x 108 IU/mL.
4. Cell Harvest and Preparation for Infusion (Day 1)
- Harvest the cells.
- Collect transduced and mock cells as described in steps 3.3.1 – 3.3.2. Perform sequential washes with 10 mL of HBSS, transferring the washes to the conical tube with the cells. Ensure that all the cells have been removed from the flasks, tapping/slapping the flasks if necessary.
- Centrifuge the cell suspensions at 800 x g for 5 min, aspirate the supernatant, and resuspend the pellet in 1 mL of HBSS. Combine the tubes containing cells from the same condition. Rinse each with 10 mL of HBSS and add that to the cell suspension. Determine the cell counts using a hemocytometer. Bring the total cell volume to 50 mL in HBSS and centrifuge at 800 x g for 5 min.
- Pulse prostaglandin E2 (PGE2) and prepare reagents for the infusion product.
- Aspirate the supernatant and resuspend the transduced cells (not mock) to 5 x 106/mL in HSPC media without cytokines. Add 10 mM PGE2 to a final concentration of 10 µM. Incubate the cells on ice for 2 h, gently swirling them every 30 min.
- During step 4.2.1, heat-inactivate autologous serum (Table of Materials), wrapping the container in wax paper and incubating it at 56 °C for 30 min. Prepare 2% autologous serum in HBSS (500 µL of heat-inactivated serum + 24.5 mL of HBSS), mix them well, and store the mixture on ice until use. Also, during step 4.2.1, harvest mock-transduced cells in a 12-well plate by pipetting the cells up and down vigorously. Add the cell suspension to a 15 mL conical tube, wash them 3x with 1 mL of HBSS, and add the washes to the same 15 mL conical tube.
- After 2 h of PGE2 incubation, wash the cells 2x by centrifuging them at 800 x g for 5 min and discarding the supernatant. After the second wash, resuspend the cells in an appropriate volume of HBSS for counting, using a hemocytometer or an automated cell counter.
NOTE: Cells frequently clump after a PGE2 pulse, which can make for less reliable cell counts. If so, use the cell count from step 4.1.2 instead of the one from step 4.2.3.
- Reserve mock and transduced cells for the quality control (step 5) of the infusion product: 5 x 105 to 1 x 106 cells for flow cytometry/cell sorting (steps 5.1 and 5.3) and approximately 1 x 106 cells for liquid culture (step 5.2) and colony polymerase chain reaction (PCR) (step 5.4).
- Prepare the infusion product.
- With the remainder of the transduced cells, perform a third wash in 50 mL of HBSS, aspirate the supernatant, and resuspend the cells in 10 mL of HBSS + 2% autologous serum (prepared in step 4.2.2). Draw the 10 mL solution into a 20 mL syringe fitted with a 16.5 G needle. Wash the tube with 10 mL of HBSS + 2% auto serum and draw the wash into the same syringe to bring the total volume to 20 mL. Cap the syringe with the needle cap, label the syringe, and place it on ice for transport/infusion.
- Infuse the transduced cell product into the autologous host, located within an accredited animal research facility, via a central venous catheter33,34. Monitor the transplanted animals’ complete blood cell counts (CBCs) and blood chemistries and perform study-specific gene-marking assays tailored to the project1,6,19.
5. Quality Control
NOTE: Flow cytometry and cell sorting are performed after the cells are infused into the animals as part of the follow-up described in step 4.4.2. Flow cytometric data is used immediately after transplantation to determine the composition of phenotypically defined stem and progenitor cell subsets in the infusion product (steps 5.1 and 5.3), whereas the analysis of CFC assays is performed 12 – 14 days postinfusion to determine the gene-modification efficiency by colony PCR (step 5.4).
- Perform flow cytometry and CFC sorting of the infusion product.
- Analyze the cells by flow cytometry and sort-purify CD34 subsets for CFC assays, as described in section 2. Seed CFC assays from mock and transduced CD34 cells (the infusion product after the PGE2 pulse).
- Sort a maximum of 800 cells per condition and vortex, plate, culture, and count CFC assays as described in step 2.4. After counting, pick the colonies from the CFC assays to perform colony PCR as described in step 5.4.
- Liquid culture
- Plate cells for liquid culture in HSPC media at 1 x 106/mL in non-TC-treated plates. On days 2, 5, and 12 posttransduction, harvest and count the cells for flow cytometry. Cell sorting for CFC assays is not required.
- On days 2 and 5, replate 33% of the cells in fresh HSPC media at 1 x 106/mL. Freeze 33% of the cells in DNA extraction buffer for quantitative real-time PCR. Use 33% of the cells for flow cytometry as described in steps 2.1 and 2.2.
- Use these data from step 5.2.2 to analyze the phenotypic composition of hematopoietic progeny. Determine the frequency of CD34+ cells and phenotypically defined subsets as described in step 2.2.2 within each sample. Compare the composition of CD34+ cells between conditions to determine the effect of gene modification.
NOTE: CD34+ cells should maintain their expression throughout the entire culture and contain all phenotypic subsets. A loss of CD90+CD45RA– cells or an overrepresentation of phenotypical subsets can indicate indirect or direct effects of gene modification.
- Quantify transgene expression (e.g., green fluorescent protein [GFP]) by flow cytometry (optional and depending on the experimental setup), adapting the protocol from step 2.2.
- Add three flow plots showing SSC/transgene (e.g., SSC/GFP). Gate the first plot on HSCs, the second on MPPs-EMPs, and the third on LMPs. Plot each versus transgene (e.g., GFP) and create gates named HSC-GFP+, MPP-EMP-GFP+, and LMP-GFP+, respectively.
- When complete, the Population hierarchy has to contain nine hierarchically organized entries in the following order: 1) All Events; 2) Scatter; 3) CD34+; 4) HSC; 5) HSC-GFP+; 6) MPP-EMP; 7) MPP-EMP-GFP+; 8) LMP; 9) LMP-GFP+.
NOTE: Any transgene expression later in liquid culture (e.g., days 5 and 12) will better reflect the gene modification efficiency determined by colony PCR in step 5.4. Earlier time points in liquid culture may overestimate the transgene expression, due to unintegrated LVs. Similarly, flow-sorting GFP+ cells for CFC assays on day 1 will result in many false positive colonies.
- Perform colony PCR.
- After the counting in steps 2.4.4 and 5.1.1, pick single colonies into 50 µL of DNA extraction buffer, using a 10 µL or 20 µL pipette, and pipette up and down to transfer the colonies to one tube of an 8-tube PCR strip cap. Pick eight colonies from the mock condition and 88 colonies from the transduced condition to generate one full 96-well plate. Add 50 µL of DNA extraction buffer to each well.
- Extract DNA from the colonies, using a thermocycler and the following program: 65 °C/20 min, 99 °C/10 min, and a 4 °C hold. Plan to move the DNA to -20 °C as soon as possible for optimal quality.
- Perform colony PCR to quantify LV-transduced cells, comparing the number of LV+ colonies, using Lenti F/R primers, to total colonies, using Actin F/R and control primers (Table of Materials).
Representative Results
The protocol described above is designed to isolate and gene-modify NHP CD34+ HSPCs, which can subsequently be infused back into the autologous host (Figure 1 and Figure 2). When following this protocol, we usually obtain up to 8 x 109 total WBCs from primed BM from pigtail macaques and, sometimes, double that amount from rhesus macaques. In both species, the number of CD34+ HSPCs that we enrich is proportional to the input of the total WBC count (Figure 3). Previous findings demonstrate that the total CD34+ HSPC product includes cells that are not true, long-term engrafting HSCs. Hence, we have developed flow-cytometry-based techniques (Figure 4) and use CFC assays (Figures 5-6) to identify long-term HSCs and committed progenitor subsets in cultures. Unlike true HSCs, committed progenitors will persist for a relatively short time period in vivo. Finally, the LV-mediated gene modification strategy results in robust gene marking in cultured CD34+ HSPCs. These cells are monitored over up to 2 weeks in culture, in part to reduce the number of "false positive" cells that express GFP from nonintegrated LV vectors. Because these cells do not carry a stably integrated copy of the LV vector in the cellular genome, they will dilute out over the 12-day liquid culture assay (Figure 7).
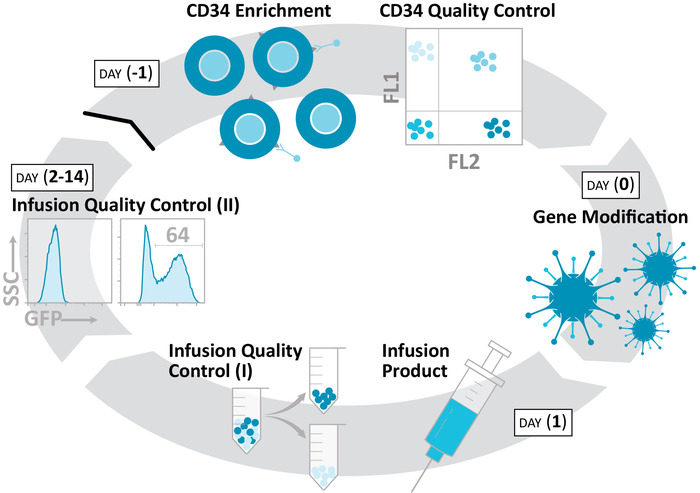
Figure 1: Production of an autologous, gene-modified NHP HSPC product. The protocol isolates CD34+ HSPCs (day -1), gene-modifies these cells (day 0), and prepares an infusion product (day 1) over a 48 h time course. Colony formation, liquid culture, and related ex vivo assays continue for an additional 2 weeks, in order to characterize these products. Please click here to view a larger version of this figure.
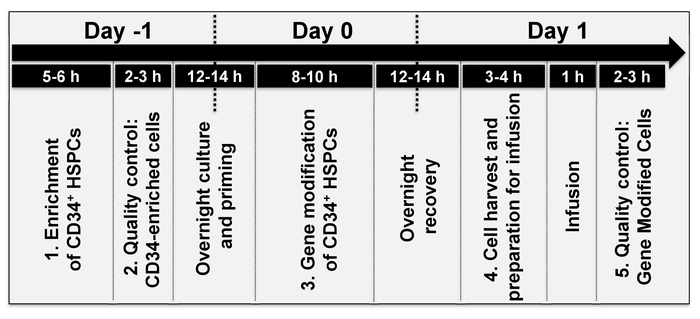
Figure 2: Timeline of events. The average time to perform each individual step of the protocol, over approximately 2 days. The timing of individual steps varies depending on the stem cell source (step 1 of the protocol) and/or the type of gene modification (step 3 of the protocol). Please click here to view a larger version of this figure.
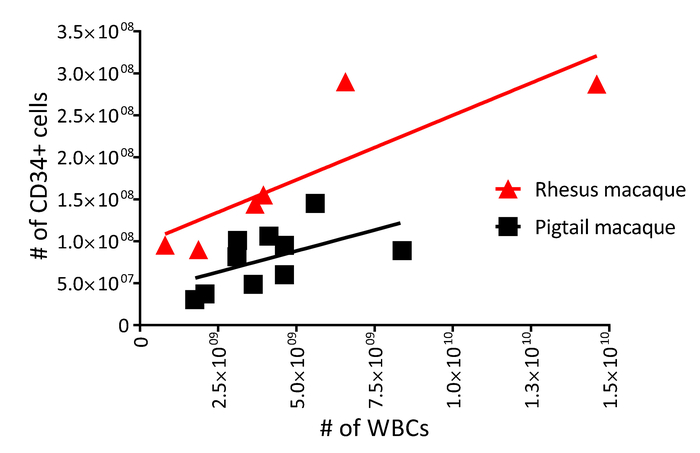
Figure 3: White blood cell and CD34 yield from primed pigtail and rhesus macaque bone marrow. Enriched CD34+ HSPC counts (step 1.4 of the protocol) as a function of total white blood cell counts (step 1.3.3 of the protocol) from 10 pigtail macaques (squares) and six rhesus macaques (triangles). Please click here to view a larger version of this figure.
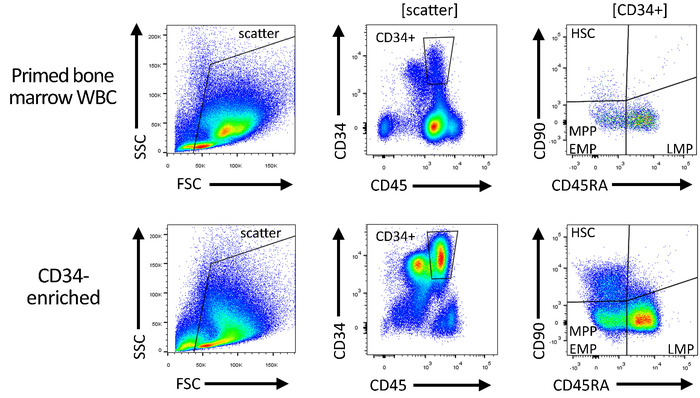
Figure 4: Gating strategy for the quality control of CD34-enriched and gene-modified products. Total white blood cells ("pre-enrichment") and subsequently CD34-enriched HSPCs are stained with antibodies specific for CD34, CD45, CD90, and CD45RA, in order to quantify the number of HSC-, MPP-, EMP-, and LMP-enriched CD34 subsets. Please click here to view a larger version of this figure.
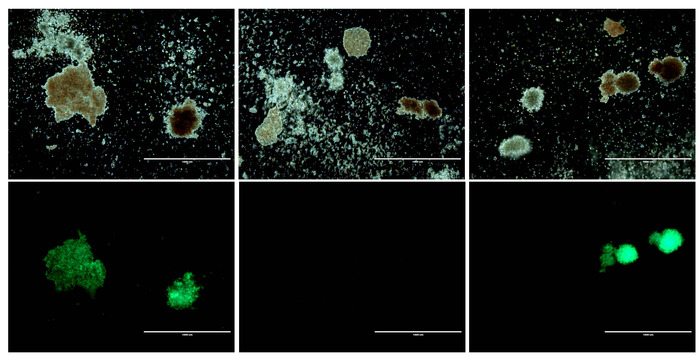
Figure 5: Morphology of CD34+ cells before and after gene modification. Top panels: Three representative brightfield images from HSPC colony assays. Bottom panels: GFP fluorescence corresponding to each brightfield image above. Scale bar = 1 mm. Please click here to view a larger version of this figure.
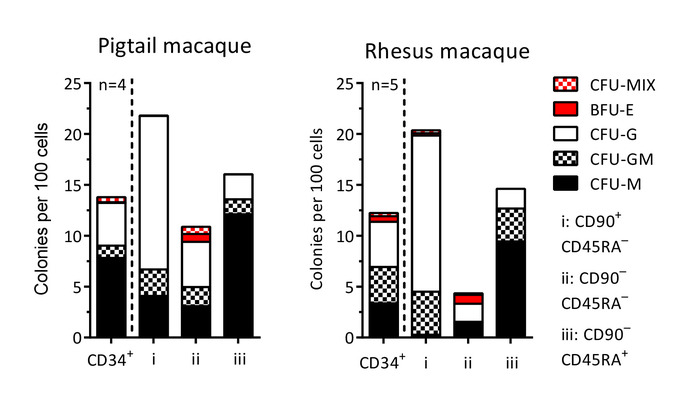
Figure 6: Colony-forming cell (CFC) potential of CD34+ cells and sort-purified subsets. Following the sort purification for CFC assays from HSPC subsets on day -1 (sections 1 and 2 of the protocol) and day 1 (section 5 of the protocol), single colonies are scored based on morphological characteristics. CFU-MIX = mix of myeloid (white) and erythroid (red) cells; BFU-E = only erythroid cells; CFU-G = granulocytes; CFU-GM = granulocytes and macrophages/monocytes; CFU-M = macrophages/monocytes. Please click here to view a larger version of this figure.
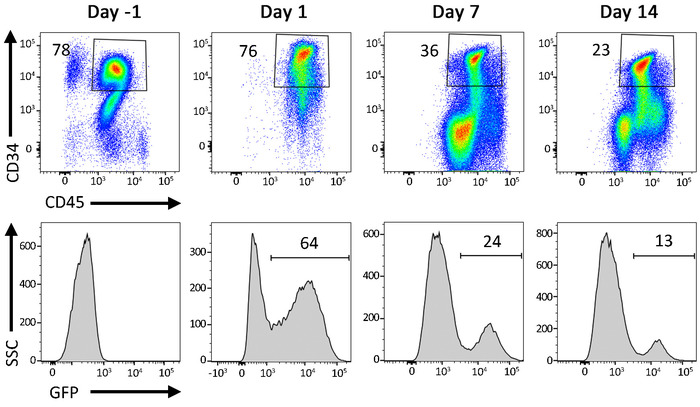
Figure 7: Representative flow cytometric data from gene-modified cells in liquid culture. Percentage of CD34+ HSPCs following transduction with a GFP-expressing LV vector. Cells are cultured for up to 2 weeks following transduction. A decrease in GFP+ events over time reflects a loss of GFP signal from cells carrying nonintegrated LV vectors. Please click here to view a larger version of this figure.
| Name | Contents |
| Hemolytic Buffer | 150 mM Ammonium Chloride, 12 mM Sodium Bicarbonate, 0.1 mM EDTA in double-distilled water (ddH2O) |
| Commercial Buffer | Phosphate-buffered saline (1X) pH 7.2, 0.5% BSA, 2 mM EDTA |
| FACS buffer | Phosphate-buffered saline (1X) pH 7.2, 2% Fetal Bovine Serum |
| HBSS + 2% BSA | Hank's Balanced Salt Solution, 2% Bovine Serum Albumin |
| HSPC Media | StemSpan SFEM II, 1% Penicillin/Streptomycin, 100 ng/mL each recombinant human TPO, SCF, FLT-3 |
| Transduction Media | StemSpan SFEM II, 1% Penicillin/Streptomycin, 100 ng/mL each recombinant human TPO, SCF, FLT-3, 1 µg/mL Cyclosporine, 4 ug/mL Protamine Sulfate |
Table 1: Buffer and media formulations.
| ID | PE | PECF594 | APC-Cy7 | V450 | Description |
| 1 | CD90 | Compensation Beads | |||
| 2 | CD34 | Compensation Beads | |||
| 3 | CD45RA | Compensation Beads | |||
| 4 | CD45 | Compensation Beads | |||
| 5 | Unstained WBCs before CD34-enrichment | ||||
| 6 | CD90 | CD34 | CD45RA | CD45 | Stained WBCs before CD34-enrichment |
| 7 | Unstained WBCs of flow-through (FT) | ||||
| 8 | CD90 | CD34 | CD45RA | CD45 | Stained WBCs of flow-through (FT) |
| 9 | Unstained WBCs of CD34-enriched product | ||||
| 10 | CD90 | CD34 | CD45RA | CD45 | Stained WBCs of CD34-enriched product |
Table 2: Representative flow panel for the quality control of CD34-enriched and gene-modified cell products.
| Antigen | Clone | Fluorochrome | Laser | Filter |
| CD45 | D058-1284 | V450 | 395 nm | 450/40 nm |
| CD90 | 5.00E+10 | PE | 488 nm or 532 nm | 585/42 nm |
| CD34 | 563 | PE-CF594 | 488 nm or 532 nm | 610/20 nm |
| CD45RA | 5H9 | APC-Cy7 | 633 nm | 780/60 nm |
| V450: violet 450 nm; PE: Phycoerythrin; PE-CF594: trademark name from Biotium; APC: Allophycocyanin-cyanine 7 | ||||
Table 3: Antibody-staining panel for quality control by flow cytometry and cell sorting.
Discussion
LV vector engineering is the best-characterized method to gene-modify cell types such as CD34+ HSPCs, for subsequent transplantation in vivo. The protocol described here is designed to maximize the number of gene-modified HSPCs that persist long-term in vivo, and provide clinical benefits to patients with various malignant, infectious, and genetic diseases. Although gene-editing strategies have emerged over the last decade, LV-modified cells are the best studied in vitro, in animal models, and in patients1,8,20,21,22.
Based on our extensive experience with this protocol, the enrichment of pure CD34+ HSPCs (i.e., >80% of the CD34+ cells in the sorted cell product) is a critical aspect of success. Because these cells are derived from a mixed population of total BM WBCs, low-purity cultures may include cells that will not engraft long-term, in turn lowering the dose of true stem cells that are infused into the autologous host. Additionally, high-quality LV VCM will ensure the highest efficiency of gene modification.
To address shortcomings in the purity of enriched CD34+ HSPC products, it is often useful to validate the quality of the reagents used to isolate these cells, namely the anti-CD34 antibody (which our group purifies in-house from a hybridoma cell line), and magnetic beads which bind antibody-labeled cells. We recommend an MOI of 10, with an option to repeat this transduction (MOI 10 x 2). Importantly, the MOI should be determined empirically, following a quality assessment of VCM, including the titration of each vector on a standardized titering cell line such as HT108029. Importantly, different VCM-producing laboratories may use distinct titering cell lines, including HOS30, HeLA31, and 293T32, which may limit the ability to compare titers between facilities. We prefer to retiter a vector with the assay, in order to calculate the most accurate amount of VCM to efficiently gene-modify CD34+ HSPC target cells. Once high-quality CD34+ HSPCs and VCM have been obtained, transduction efficiency and engraftment are further enhanced in three distinct steps. First, the addition of cyclosporine to transduction media aids in the early steps of vector transduction and integration into target cells35,36, while protamine sulfate decreases repulsive forces between the lentiviral vector particles and the cell surface33,37. Second, treating plates with a recombinant fibronectin fragment (e.g., RetroNectin, CH-296) increases transduction efficiency and also improves the in vivo engraftment potential of gene-modified HSPCs 33,38,39. Notably, previous studies suggest that CH-296 is only important during, but not prior to, transduction33,38. Finally, we pulse gene-modified cell products with PGE2 to enhance engraftment and persistence in vivo, as has previously been shown for human and nonhuman primate CD34+ HSPC products40,41.
LVs integrates randomly into the genome, as do their predecessor, gammaretroviral vectors (RVs). Although less clinical trial data is available for LVs than for RVs, current findings suggest that LV approaches carry substantially lower risks of cell transformations due to LV's insertional mutagenesis; RV strategies are less often used due to this risk42. A further limitation is that the efficiency of gene modification is less than 100%, and a proportion of gene-modified cells will not persist in vivo. For example, a 60% gene-modified HSPC product may result in 30% long-term engrafting, gene-modified cells. Whenever possible, the gene therapy strategies presented here are designed to overcome this limitation by introducing transgenes that provide therapeutic efficacy, even when expressed in a minority of hematopoietic-origin cells8,43.
The future is bright for LV-based HSPC modification approaches. We routinely use this protocol to "gene-mark" cells, enabling tracking in vivo1. We have also adapted this strategy to compare LV variants within the same animal. Such "competitive" transplants allow a comparison of different vectors (e.g., to identify those with better efficiency) and transgenes (e.g., to track different HSPC subsets or compare them in disease models). Moving forward, we believe that LV gene therapy in HSPCs will remain an essential tool alongside gene-editing approaches, especially in situations where large genetic cargoes must be stably expressed for the lifetime of an individual.
Offenlegungen
The authors have nothing to disclose.
Acknowledgements
The authors thank Helen Crawford for preparing this manuscript, Jim Woolace for graphic design, and Veronica Nelson and Devikha Chandrasekaran for participating in the development of the protocol. The development of this protocol was supported by grants from the NIH National Institute of Allergy and Infectious Diseases (R01 AI135953 and AI138329 to H.P.K.) and the National Heart, Lung, and Blood Institute (R01 HL136135, HL116217, P01 HL122173, and U19 HL129902 to H.P.K.), as well as NIH P51 OD010425 and, in part through the NIH/NCI Cancer Center, Support Grant P30 CA015704. H.P.K. is a Markey Molecular Medicine Investigator and received support as the inaugural recipient of the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research and the Fred Hutch Endowed Chair for Cell and Gene Therapy.
Materials
| Stemspam SFEM II ("HSPC") Media | StemCell | 09655 | |
| Hank's Balanced Salt Solution | Gibco | 14175095 | |
| Phosphate-Buffered Saline | Gibco | 14190-144 | |
| Penicillin/Streptomycin | Gibco | 15140-122 | |
| Dimethyl Sulfoxide | Sigma Aldrich | D2650-100 | |
| 100% Ethanol | Decon labs | M18027161M | |
| Cyclosporine | Sigma | 30024-25MG | |
| 500 mM EDTA | Invitrogen | 15575-038 | |
| Heat-Inactivated Fetal Bovine Serum | Sigma Aldrich | PS-0500-A | |
| CH-296/ RetroNectin (2.5 mL, 1 µg/µL) | TaKaRA | T100B | |
| Bovine Serum Albumin | Sigma | A7906-100g | |
| HEPES | Sigma | H9897 | |
| Rat anti-mouse IgM magnetic beads | Miltenyi Biotec | 130-047-301 | |
| Recombinant HumanStem Cell Factor (SCF) | Peprotech | 300-07 | |
| Recombinant Human Thrombopoietin (TPO) | Peprotech | 300-18 | |
| Recombinant Human FMS-like tyrosine kinase 3 (FLT-3) | Peprotech | 300-19 | |
| Protamine sulfate | Sigma | P-4505 | |
| 14 mL Polypropylene Round-Bottom Tube | Corning | 352059 | |
| Colony Gel 1402 | ReachBio | 1402 | |
| QuadroMACS Separators | Miltenyi Biotec | 130-090-976 | |
| MACS L25 Columns | Miltenyi biotec | 130-042-401 | |
| 10 mM PGE2 | Cayman Chemical | 14753-5mg | |
| TC-treated T-75 flasks | Bioexpress | T-3001-2 | |
| Non-TC-treated T-75 flasks | Thermo-Fisher | 13680-57 | |
| 20 ml syringes | BD Biosciences | 302830 | |
| 16.5 G needles | BD Precision | 305198 | |
| Syringe Tip Cap | BD Biosciences | 305819 | |
| QuickExtract DNA Solution | Epicentre | QE09050 | |
| 8-tube strip cap PCR Tubes | USA scientific | 1402-2708 | |
| 96-well Thermocycler | Thermo-Fisher | 4375786 | |
| Pre-Separation filters | Miltenyi Biotec | 130-041-407 | |
| Strainer, Cell; BD Falcon; Sterile; Nylon mesh; Mesh size: 70um; Color: white; 50/CS | fisher scientific | 352350 | |
| Ultracomp ebeads | eBioscience | 01-2222-42 | |
| MACSmix Tube Rotator | Miltenyi | 130-090-753 | |
| 3 mL Luer-Lock Syringes | Thermo-Fisher | 14823435 | |
| 35 mm x 10 mm cell culture dish | Corning | 430165 | |
| 60 mm x 15 mm cell culture dish | Corning | 430196 | |
| 150 mm x 25 mm cell culture dish | Corning | 430599 | |
| Non TC treated flasks | Falcon | 353133 | |
| Qiagen DNA extraction | Qiagen | 51104 | |
| PE Anti-Human CD90 (Thy1) Clone:5E10 | Biolegend | 328110 | |
| PE-CF594 Mouse Anti-Human CD34 Clone:563 | BD horizon | 562449 | |
| APC-H7 Mouse Anti-Human CD45RA Clone: 5H9 | BD Pharmingen | 561212 | |
| V450 Mouse Anti-NHP CD45 Clone:d058-1283 | BD Biosciences | 561291 | |
| Autologous Serum | Collected from autologous host and cryopreserved prior to mobilization and collection of CD34+ HSPCs | N/A | Beard, B. C. et al. Efficient and stable MGMT-mediated selection of long-term repopulating stem cells in nonhuman primates. Journal of Clinical Investigation. 120 (7), 2345-2354, (2010). |
| Virus-Conditioned Media (VCM) | Kiem Lab, FHCRC Co-operative Center for Excellence in Hematology (CCEH) | N/A | Beard, B. C. et al. Efficient and stable MGMT-mediated selection of long-term repopulating stem cells in nonhuman primates. Journal of Clinical Investigation. 120 (7), 2345-2354, (2010). |
| Anti-CD34 antibody, Clone 12.8 | Kiem Lab | N/A | Beard, B. C. et al. Efficient and stable MGMT-mediated selection of long-term repopulating stem cells in nonhuman primates. Journal of Clinical Investigation. 120 (7), 2345-2354, (2010). |
| Lenti F primer: AGAGATGGGTGCGAGAGCGTCA | Integrated DNA Technologies | N/A | Peterson, C. W. et al. Multilineage polyclonal engraftment of Cal-1 gene-modified cells and in vivo selection after SHIV infection in a nonhuman primate model of AIDS. Mol Ther Methods Clin Dev. 3 16007, (2016). |
| Lenti R primer: TGCCTTGGTGGGTGCTACTCCTAA | Integrated DNA Technologies | N/A | Peterson, C. W. et al. Multilineage polyclonal engraftment of Cal-1 gene-modified cells and in vivo selection after SHIV infection in a nonhuman primate model of AIDS. Mol Ther Methods Clin Dev. 3 16007, (2016). |
| Actin F primer: TCCTGTGGCACTCACGAAACT | Integrated DNA Technologies | N/A | Peterson, C. W. et al. Multilineage polyclonal engraftment of Cal-1 gene-modified cells and in vivo selection after SHIV infection in a nonhuman primate model of AIDS. Mol Ther Methods Clin Dev. 3 16007, (2016). |
| Actin R primer: GAAGCATTTGCGGTGGACGAT | Integrated DNA Technologies | N/A | Peterson, C. W. et al. Multilineage polyclonal engraftment of Cal-1 gene-modified cells and in vivo selection after SHIV infection in a nonhuman primate model of AIDS. Mol Ther Methods Clin Dev. 3 16007, (2016). |
Referenzen
- Radtke, S., et al. A distinct hematopoietic stem cell population for rapid multilineage engraftment in nonhuman primates. Science Translational Medicine. 9 (414), (2017).
- Majeti, R., Park, C. Y., Weissman, I. L. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 1 (6), 635-645 (2007).
- Notta, F., et al. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science. 333 (6039), 218-221 (2011).
- Hoggatt, J., et al. Rapid mobilization reveals a highly engraftable hematopoietic stem cell. Cell. 172 (1-2), 191-204 (2018).
- Chandrasekaran, D., Nakamoto, B., Watts, K. L., Kiem, H. P., Papayannopoulou, T. Modeling promising nonmyeloablative conditioning regimens in nonhuman primates. Human Gene Therapy. 25 (12), 1013-1022 (2014).
- Humbert, O., Peterson, C. W., Norgaard, Z. K., Radtke, S., Kiem, H. P. A nonhuman primate transplantation model to evaluate hematopoietic stem cell gene editing strategies for beta-hemoglobinopathies. Molecular Therapy. Methods & Clinical Development. 8, 75-86 (2018).
- Peterson, C. W., et al. Differential impact of transplantation on peripheral and tissue-associated viral reservoirs: Implications for HIV gene therapy. PLoS Pathogens. 14 (4), e1006956-e1006956 (2018).
- Zhen, A., et al. Long-term persistence and function of hematopoietic stem cell-derived chimeric antigen receptor T cells in a nonhuman primate model of HIV/AIDS. PLoS Pathogens. 13 (12), e1006753 (2017).
- Moffett, H. F., et al. Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nature Communications. 8 (1), 389 (2017).
- Burtner, C. R., et al. Intravenous injection of a foamy virus vector to correct canine SCID-X1. Blood. 123 (23), 3578-3584 (2014).
- Evans, D. T., Silvestri, G. Nonhuman primate models in AIDS research. Current Opinion in HIV and AIDS. 8 (4), 255-261 (2013).
- Taraseviciute, A., et al. Chimeric antigen receptor T cell-mediated neurotoxicity in nonhuman primates. Cancer Discovery. 8 (6), 750-763 (2018).
- McGary, C. S., et al. CTLA-4(+)PD-1(-) memory CD4(+) T cells critically contribute to viral persistence in antiretroviral therapy-suppressed, SIV-infected rhesus macaques. Immunity. 47 (4), 776-788 (2017).
- Beard, B. C., Adair, J. E., Trobridge, G. D., Kiem, H. P. High-throughput genomic mapping of vector integration sites in gene therapy studies. Methods in Molecular Biology. 1185, 321-344 (2014).
- Zonari, E., et al. Efficient ex vivo engineering and expansion of highly purified human hematopoietic stem and progenitor cell populations for gene therapy. Stem Cell Reports. 8 (4), 977-990 (2017).
- Doulatov, S., et al. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nature Immunology. 11 (7), 585-593 (2010).
- Fares, I., et al. Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science. 345 (6203), 1509-1512 (2014).
- Gori, J. L., et al. Endothelial cells promote expansion of long-term engrafting marrow hematopoietic stem and progenitor cells in primates. Stem Cells Translational Medicine. 6 (3), 864-876 (2017).
- Peterson, C. W., et al. Long-term multilineage engraftment of autologous genome-edited hematopoietic stem cells in nonhuman primates. Blood. 127 (20), 2416-2426 (2016).
- Peterson, C. W., et al. Multilineage polyclonal engraftment of Cal-1 gene-modified cells and in vivo selection after SHIV infection in a nonhuman primate model of AIDS. Molecular Therapy. Methods & Clinical Development. 3, 16007 (2016).
- Thompson, A. A., et al. Gene therapy in patients with transfusion-dependent beta-thalassemia. The New England Journal of Medicine. 378 (16), 1479-1493 (2018).
- Eichler, F., et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. The New England Journal of Medicine. 377 (17), 1630-1638 (2017).
- Paul, B., et al. Efficient enrichment of gene-modified primary T cells via CCR5-targeted integration of mutant dihydrofolate reductase. Molecular Therapy. Methods & Clinical Development. 5 (9), 347-357 (2018).
- Levine, B. L., Miskin, J., Wonnacott, K. Global manufacturing of CAR T cell therapy. Molecular Therapy. Methods & Clinical Development. 4, 92-101 (2017).
- Masiuk, K. E., et al. Improving gene therapy efficiency through the enrichment of human hematopoietic stem cells. Molecular Therapy. 25 (9), 2163-2175 (2017).
- Trobridge, G. D., et al. Efficient transduction of pigtailed macaque hematopoietic repopulating cells with HIV-based lentiviral vectors. Blood. 111 (12), 5537-5543 (2008).
- Beard, B. C., et al. Efficient and stable MGMT-mediated selection of long-term repopulating stem cells in nonhuman primates. Journal of Clinical Investigation. 120 (7), 2345-2354 (2010).
- Horn, P. A., et al. Efficient lentiviral gene transfer to canine repopulating cells using an overnight transduction protocol. Blood. 103 (10), 3710-3716 (2004).
- Adair, J. E., et al. Semi-automated closed system manufacturing of lentivirus gene-modified haematopoietic stem cells for gene therapy. Nature Communications. 7, 13173 (2016).
- Kutner, R. H., Zhang, X. Y., Reiser, J. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nature Protocols. 4 (4), 495-505 (2009).
- Salmon, P., Trono, D. Production and titration of lentiviral vectors. Current Protocols in Human Genetics. 54 (1), (2007).
- Geraerts, M., Willems, S., Baekelandt, V., Debyser, Z., Gijsbers, R. Comparison of lentiviral vector titration methods. BMC Biotechnology. 6, 34 (2006).
- Kiem, H. P., et al. Improved gene transfer into baboon marrow repopulating cells using recombinant human fibronectin fragment CH-296 in combination with interleukin-6, stem cell factor, FLT-3 ligand, and megakaryocyte growth and development factor. Blood. 92 (6), 1878-1886 (1998).
- Morton, W. R., Knitter, G. H., Smith, P. M., Susor, T. G., Schmitt, K. Alternatives to chronic restraint of nonhuman primates. Journal of the American Veterinary Medical Association. 191 (10), 1282-1286 (1987).
- Noser, J. A., et al. Cyclosporine increases human immunodeficiency virus type 1 vector transduction of primary mouse cells. Journal of Virology. 80 (15), 7769-7774 (2006).
- Uchida, N., Hsieh, M. M., Washington, K. N., Tisdale, J. F. Efficient transduction of human hematopoietic repopulating cells with a chimeric HIV1-based vector including SIV capsid. Experimental Hematology. 41 (9), 779-788 (2013).
- Cornetta, K., Anderson, W. F. Protamine sulfate as an effective alternative to polybrene in retroviral-mediated gene-transfer: implications for human gene therapy. Journal of Virological Methods. 23 (2), 187-194 (1989).
- Goerner, M., et al. The use of granulocyte colony-stimulating factor during retroviral transduction on fibronectin fragment CH-296 enhances gene transfer into hematopoietic repopulating cells in dogs. Blood. 94 (7), 2287-2292 (1999).
- Dao, M. A., Hashino, K., Kato, I., Nolta, J. A. Adhesion to fibronectin maintains regenerative capacity during ex vivo culture and transduction of human hematopoietic stem and progenitor cells. Blood. 92 (12), 4612-4621 (1998).
- North, T. E., et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 447 (7147), 1007-1011 (2007).
- Goessling, W., et al. Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell. 8 (4), 445-458 (2011).
- Booth, C., Gaspar, H. B., Thrasher, A. J. Treating immunodeficiency through HSC gene therapy. Trends in Molecular Medicine. 22 (4), 317-327 (2016).
- Humbert, O., et al. Rapid immune reconstitution of SCID-X1 canines after G-CSF/AMD3100 mobilization and in vivo gene therapy. Blood Advances. 2 (9), 987-999 (2018).
