Nuclear dUTPase forms an intermolecular disulfide linkage forming a stable dimer configuration through the interaction of two cysteine residues positioned at the third amino acid of each monomeric protein11. This is demonstrated in Figure 1A,B. To ensure this disulfide linkage was not a nonspecific interaction due to migration abnormalities in the non-reduced environment,t the inclusion of a proper control was essential. Of note, nuclear dUTPase is one of four isoforms present in humans. Three of the four isoforms have a unique amino-terminal domain while sharing a common catalytic core11,12. As seen on the western blot, the monomeric confirmation of dUTPase in human cells at the time of harvesting is a combination of at least three of the isoforms of dUTPase (the mitochondria isoform, the nuclear isoform, and a truncated version notated at M24), all of which are recognized by our polyclonal antibody. The nuclear isoform is the only isoform that contains a cysteine residue in its unique amino terminal domain. Due to the nuclear isoform being only a small percentage of the monomeric state of the proteins seen on the western blot, it was exposed longer to demonstrate the dimeric state of that isoform.
The mitochondrial isoform lacks the cysteine residue present in nuclear isoform and was used as a control for this intermolecular disulfide linkage. As seen in Figure 1C, isolation of mitochondria followed by a western blot analysis demonstrated that this isoform under non-reducing conditions did not form a disulfide linkage and migrated to the predicted molecular weight for the monomeric protein.
To confirm this complex can form a multimeric complex, formaldehyde cross-linking was performed. Isolated nuclei were subjected to 1% formaldehyde treatment followed by denaturing SDS-PAGE/western blot analysis under reducing conditions. As demonstrated in Figure 2, the dimerization was visualized. When the cross-link was reversed by incubating the sample at 95 °C for 15 min, the complex was destabilized and could be visualized in its monomeric state.
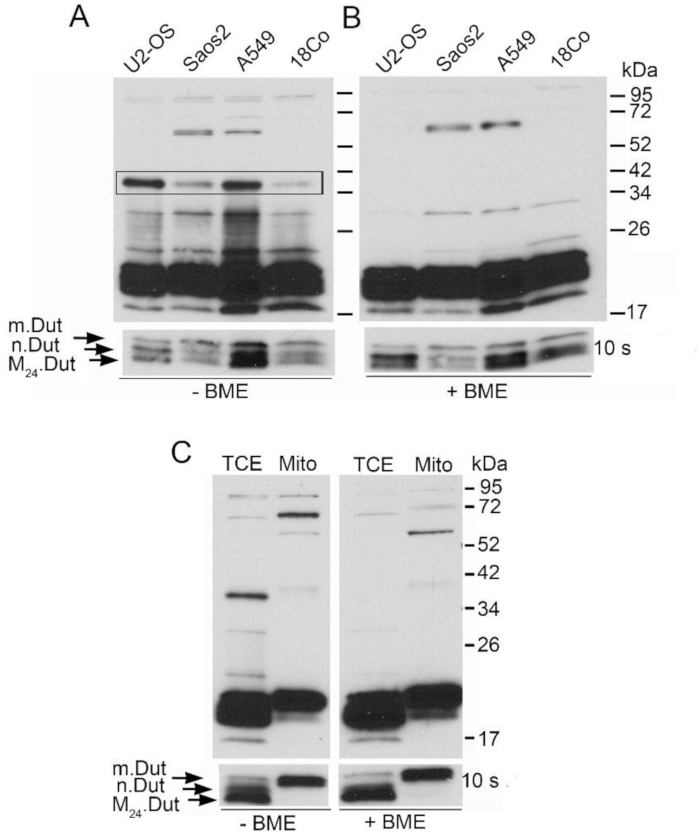
Figure 1: Demonstration of intermolecular disulfide bond formation in the nuclear dUTPase protein. (A) A western blot analysis of total cell extracts (TCE) in the absence of the reducing agent, beta-mercaptoethanol (BME), demonstrates a multimeric complex formation in asynchronous populations of U-2 OS, Saos2, A549, and 18CO as indicated by the black box. (B) This complex disappears with the addition of BME in all four cell lines examined, indicating the presence of a disulfide linkage. (C) A western blot analysis of TCE and purified mitochondrial extracts (Mito) derived from U-2 OS cells, ±BME, shows no multimeric complex formation in the -BME sample. The lower panels in panels A, B, and C demonstrate exposure to X-ray film for 10 s, showing the monomeric state of the three isoforms of dUTPase. The upper panels in A and B were exposed for 1 min, while the upper panel in C was exposed for 2 min. Equivalent amounts of protein were applied to each lane. Blots were probed with a polyclonal specific antibody against the conserved carboxyl-terminal domain of dUTPase. This figure has been modified from Rotoli et al.11 Please click here to view a larger version of this figure.
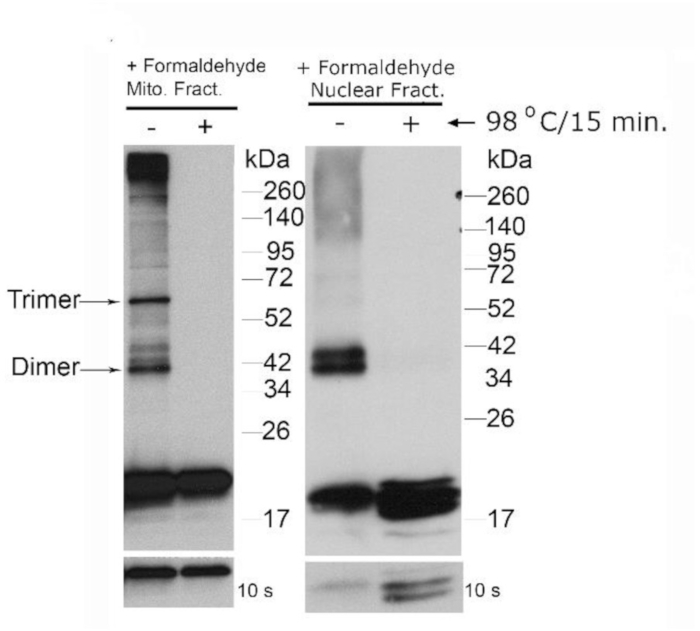
Figure 2: Formaldehyde cross-linking of nuclear dUTPase demonstrate multimeric complex formation. U-2 OS cells were incubated with 1% formaldehyde for 15 min. Nuclei (N) were isolated then analyzed by western blot using a specific polyclonal antibody against the conserved carboxyl-terminal domain of dUTPase (+ formaldehyde). To reverse the formaldehyde cross-links, extracts derived from the nuclear preparations were mixed with SDS-PAGE buffer then heated to 98 °C for 15 min in the presence of BME (+formaldehyde, 98 °C for 15 min). The observed heterogeneity (i.e. doublet bands of nDut) seen with the preparations remain to be explained, but may be due to anomalous migration due to the formaldehyde treatment. The lower panel is exposed to X-ray film for 10 s, while the upper panel is exposed to X-ray film for 2 min. This figure has been modified from Rotoli et al.11 Please click here to view a larger version of this figure.
