Our extraction method allowed us to harvest up to ~80,000 LSK HSPCs per mouse. The viability and numbers of LSK cells were improved with our method, because we: (1) combined bone marrow from upper and lower limbs, hip bones, sternum, rib cage, and spine, (2) avoided using red cell lysis buffer that would have increased cell-death and clumping, (3) used the density gradient medium separation of mono-nucleated cells, and (4) avoided using pre-chilled buffer that would have caused the loss of cells-of-interest in clumps.
Although extracellular flux analysis has been traditionally used for adherent cells, our use of the PLL coating of wells, followed by centrifugation of cells on it, facilitated adherence of LSK HSPCs to the surface of the well. This allowed us to measure the extracellular flux, and thus metabolic health of LSK HSPCs. Considering the limited number of cells that can be harvested from a mouse and the long duration of the protocol for their isolation, our use of the analyzer with its 8 well format has emerged as the most cost-effective and feasible solution (Figure 1).
Cells use glycolysis and mitochondrial respiration to replenish their energy requirements and to produce intermediates needed for their proliferation and growth19. The hexokinase enzyme converts glucose in glucose-6-phosphate and that is subsequently transformed into pyruvate20. Pyruvate can then be processed into lactate and is exported from the cell with protons21. ECAR measures the acidification of the media and is thus an indicator of glycolysis. Pyruvate can also be transported into the mitochondria and transformed in acetyl coenzyme A (CoA). Acetyl CoA enters the TCA cycle, which provides energy intermediates to drive the electron movements of the Electron Transport Chain (ETC) and generates a proton gradient in the mitochondrial inter-membrane space22. Oxygen acts as the final electron acceptor, and protons move back to the mitochondrial matrix through the ATP synthase complex while generating ATP23. OCR measures the oxygen consumption and it is therefore used to quantify the mitochondrial respiration.
In order to analyze the OCR and the ECAR in basal and stressed conditions, we used sequential injection of drugs that interfere with glycolysis and mitochondrial respiration. We used glucose and 2-deoxyglucose (2-DG), a glucose analogue, to initiate and block glycolysis respectively24. We used Rotenone (a complex I-specific inhibitor of the ETC), antimycin A (a complex III-specific inhibitor of the ETC), oligomycin (inhibitor of ATP synthase), and the uncoupling agent carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) to block specific events of the ETC25. We titrated such reagents to find the optimal concentration for LKS HSPCs (Figure 2A,B).
To perform the glycolysis stress test, we cultured the LSK HSPCs in a glucose/pyruvate deprived media (as recommended by the manufacturer), or in glucose/pyruvate containing media. As expected, we found the basal level of the ECAR was higher for LSK HSPCs cultured in glucose/pyruvate+ media compared to LSK HSPCs cultured in glucose/pyruvate- media. The first injection with glucose did not change the basal level of ECAR for LSK HSPCs cultured in glucose/pyruvate+ media while it boosted glycolysis in LSK HSPCs cultured in glucose/pyruvate- media. However, the basal level of the ECAR, after the injection with glucose, remained lower compared to the glucose/pyruvate+ group. The second injection with oligomycin, which could block the production of ATP through oxidative phosphorylation, activated the glycolysis at its maximum level of LSK HSPCs in glucose/pyruvate+ media, but it did not affect the glucose/pyruvate- group. The last injection with the glucose analogue 2-DG returned the ECAR to its non-glycolytic level (Figure 2C).
For the mitochondrial stress test, we measured the basal level of OCR of LSK HSPCs in glucose/pyruvate+ media. We first injected oligomycin that initially hyperpolarized the mitochondrial membrane, prevented more proton pumping through the ETC complexes, and thus reduced the rate of mitochondrial respiration. The second injection of FCCP ionophores pushed ETC and OCR levels to their maximum as cells tried to recover the mitochondrial membrane potential. The final injection with other two of the ETC inhibitors (antimycin A and rotenone) caused the complete stop of mitochondrial respiration and thus the OCR reverted to its minimum level (Figure 2D).
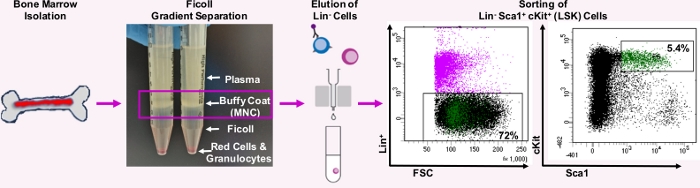
Figure 1: Schema demonstrating isolation of LineagenegSca1+c-Kit+ (LSK) hematopoietic stem progenitor cells from mouse bone marrow. Bone marrow is extracted from bones and mononuclear cells (MNCs) are isolated through density gradient medium gradient separation. Next, cells are incubated with biotinylated Lineage+ antibodies and streptavidin-conjugated magnetic beads to elute Lineage negative (Lin–) cells following their magnetic separation. Lin– cells are subsequently incubated with LSK antibodies and LSK cells isolated by cell sorting. Please click here to view a larger version of this figure.
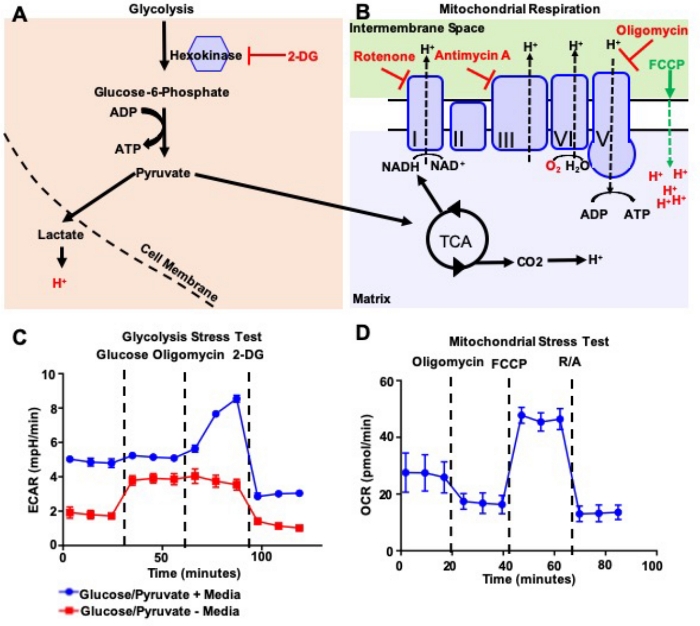
Figure 2: Extracellular flux analyses of murine LineagenegSca1+c-Kit+ (LSK) hematopoietic stem progenitor cells. (A,B) Mechanistic description of drugs utilized for extracellular flux analyses during glycolysis and mitochondrial respiration. (C) Representative results of glycolysis stress test on murine LSK HSPCs in presence or absence of glucose/pyruvate in the media. (D) Representative results of mitochondrial stress test on murine LSK HSPCs. Error bars represent the standard deviation of the mean (S.D.) Please click here to view a larger version of this figure.
| Stock | Final concentration | Volume for 30 mL | |
| Complete media | 28.691 mL | ||
| P/S | 100x | 0.5x | 150 µL |
| L-Glutamine | 200 mM | 2 mM | 300 µL |
| Pyruvate | 100 mM | 1 mM | 300 µL |
| Glucose | 1 M/180.2 mg/mL | 3 mg/mL | 499.4 µL |
| TPO | 100 µg/mL | 100 ng/mL | 30 µL |
| SCF | 100 µg/mL | 100 ng/mL | 30 µL |
Table 1: Contents and preparation of the complete XF media.
| Basal | Oligomycin (2 µM) | FCCP (1.5 µM) | R/A (0.5 µM) | |
| Repetition | 3 times | 3 times | 3 times | 3 times |
| Mix | 3 min | 3 min | 3 min | 3 min |
| Wait | 0 min | 0 min | 0 min | 0 min |
| Measure | 4 min | 4 min | 4 min | 4 min |
Table 2: Injection protocol for the mitochondrial stress test.
| Basal | Glucose (10 mM) | Oligomycin (2 µM) | 2-DG (50 mM) | |
| Repetition | 3 times | 3 times | 3 times | 3 times |
| Mix | 3 min | 3 min | 3 min | 3 min |
| Wait | 0 min | 0 min | 0 min | 0 min |
| Measure | 7 min | 7 min | 7 min | 7 min |
Table 3: Injection protocol for the glycolysis stress test.
| Port | Drug | Final well concentration (µM) | Stock solution volume (µL) | Media volume (µL) | Port solution (µM) | Volume added to port (µL) |
| Port A | Oligomycin | 2 | 100 | 181.25 | 16 | 25 |
| Port B | FCCP | 1.5 | 100 | 270.4 | 13.5 | 25 |
| Port C | Rotenone/antimycin A | 0.5 | 60 | 240 | 5 | 25 |
Table 4: Dilution metrics to obtain optimum drug concentration for mitochondrial stress test in each well of the analyzer.
| Port | Drug | Final well concentration | Stock solution volume (µL) | Media volume (µL) | Port solution | Volume added to port (µL) |
| Port A | Glucose | 10 mM | 300 | 75 | 80 mM | 25 |
| Port B | Oligomycin | 2 µM | 108 | 192 | 18 µM | 25 |
| Port C | 2-DG | 50 mM | 300 | 0 | 500 mM | 25 |
Table 5: Dilution metrics to obtain optimum drug concentration for glycolysis stress test in each well of the analyzer.
