The protocol shows how to accurately monitor the formation of aggregated species in living C. elegans, both during its natural aging and when subjected to stress. We selected four different strains of transgenic nematodes expressing polyglutamine proteins of either 40Q, 44Q, or 85Q repeats. These proteins are synthesized in different tissues and were fused to different fluorophores. The C. elegans strains either expressed Q40-mRFP in the body wall muscles (mQ40-RFP), Q40-CFP in the nervous system (nQ40-CFP), and either Q44-YFP or Q85-YFP in the intestine (iQ44-YFP and iQ85-YFP)13. To illustrate how aging promotes aggregation, we collected the lifetime of these polyQ strains in young nematodes, at day 4 of life, and old nematodes, at day 8. To show the effects of a deficiency in the PN, we performed a knockdown of hsp-1 in the mQ40-RFP and the nQ40 strains.
Once the lifetime values were extrapolated via the FLIMfit software, the obtained data showed a clear reduction in the lifetime of any of the polyQ constructs when aggregated due to either glutamine load, aging, or stress. FLIM distinguished between the soluble protein fraction and aggregated species, and their transition, by recording a shift in their lifetimes.
At day 4, mQ40-RFP displayed an average fluorescence lifetime of 1.69 ns (Figure 2). Upon aging, more aggregated species arose, appearing as low lifetime foci in the lifetime images and shifting the histogram to reduced lifetimes (Figure 2A). By plotting the mean fluorescence lifetime of every acquired image over the age of the nematodes a significant reduction of fluorescence lifetime, and therefore accumulation of aggregated species, became visible (Figure 2B). The protein folding capacity of the PN declined after day 4 of life in C. elegans14 and aggregation-prone proteins further misfolded to cluster into amyloid and amorphous aggregates. Apart from the PN, the intrinsic aggregation propensities of a certain protein played an important role in the progression of aggregate formation. This was analyzed by comparing the behavior of iQ44-YFP and iQ85-YFP. The longer Q-stretch of the iQ85 was more prone to aggregation and exhibited a fluorescence lifetime shift in the histogram already at day 4 of life (Figure 3A). In fact, at day 4, foci formation was observed for iQ85, while still absent in iQ44. Upon aging, however, iQ44 also exhibited foci formation and thus a reduced fluorescence lifetime. Because iQ85 already exhibited aggregates in early adulthood the progression of aggregation upon aging was less pronounced, yet significant (Figure 3B). Finally, we did not detect foci formation nor decreased fluorescence lifetime in the nQ40-CFP strain (Figure 4A). For this strain, there were only subtle, nonsignificant changes to the mean fluorescence lifetime upon aging (Figure 4B), potentially due to the neurons being less susceptible for yet unknown reasons.
Knocking down hsp-1 poses a challenge to the PN of mQ40 and nQ40 expressing nematodes. RNAi-mediated depletion of hsp-1 led to a significant increase in aggregation (Figure 5 and Figure 6). Q40 expressed in body wall muscles tended to form a small number of large foci surrounded by nonaggregated material. This resulted in two distinguishable peaks in the histograms (around 1.7 ns and 1.4 ns, see Figure 5A). The aged and RNAi treated nematodes showed a strong increase in the low lifetime peak ultimately decreasing the average fluorescence lifetime (Figure 5B). Compared to this biphasic behavior of Q40 in muscles, the neuronal Q40 displayed a more diverse aggregation behavior. We could not directly correlate foci formation with aggregation as in the muscular expression strain (Figure 6A). Because FLIM offers an opportunity to assess the degree of aggregation, the histograms revealed that there was no distinct peak but a widespread distribution of fluorescence lifetimes, pointing to a complex composition of different oligomers and higher order aggregates. Still, the overall degree of aggregation could be evaluated by plotting the mean fluorescence lifetime (Figure 6B), showing that hsp-1 knockdown led to a boost in aggregation.
It is important to note that the lifetime of the fluorophores, free from a fusion partner and outside of a biological system, was higher. Because the lifetime is affected primarily by its environment, a slight reduction of the lifetime of YFP and RFP was already noticeable within C. elegans' tissues. It is therefore important to obtain the lifetime of the soluble POI within the nematode as a suitable control. A comparison between the soluble fraction with a higher lifetime and aggregated fraction with a lower lifetime can then be made. Here, the decrease in lifetime correlated with the formation of visible foci within the muscle and intestinal cells. Still, a fraction of foci exhibited no decrease of fluorescence lifetime (see Figure 2 and Figure 3, white arrows). This feature highlights how only part of the fusion construct might be aggregated at a particular spatiotemporal point, and the presence and availability of unbound protein. A more complex scenario arose from investigation of the neuronal Q40-CFP strain. CFP intrinsically possesses two distinct fluorescence lifetimes. While CFP is an ideal fluorophore for Förster resonance energy transfer (FRET)15 measurements, in conjunction with YFP, it is not advisable to employ it to monitor formation of aggregates in C. elegans.
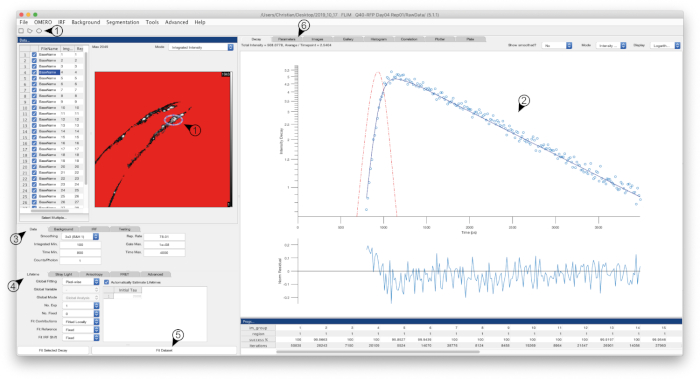
Figure 1: Screenshot of FLIMFit software interface. Screenshot of the software used to calculate the fluorescence lifetimes. The window depicts the interface after settings were defined as described in the text, and calculation of lifetimes was performed. Numbered arrows refer to specific steps within the protocol. Please click here to view a larger version of this figure.
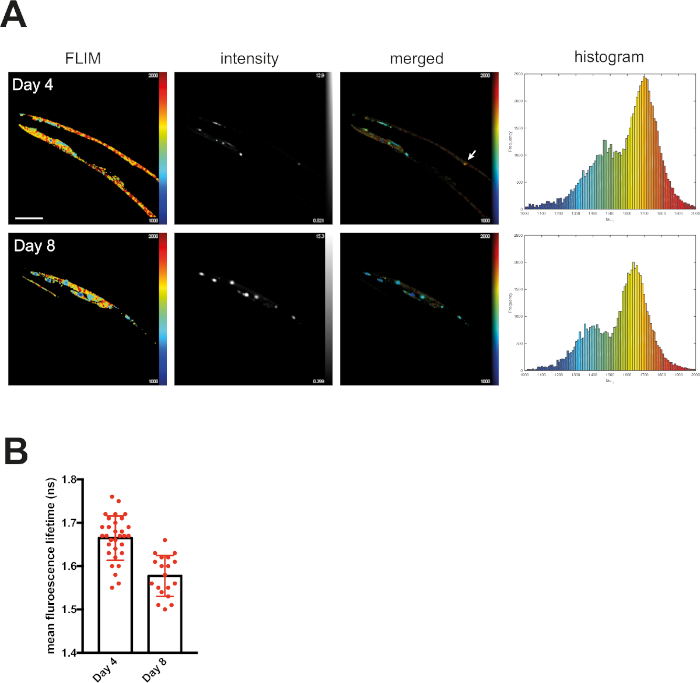
Figure 2: Fluorescence lifetimes of muscular Q40-RFP decreased with age. (A) Representative maps of C. elegans expressing muscular Q40-RFP on day 4 or day 8 of life generated by FLIMfit. Fluorescence lifetimes, fluorescence intensity, and a merged image of both are provided. Scale bars = 25 µm. Histograms show a distribution of measured lifetimes for all analyzed nematodes divided into 100 categories. (B) Bar plots showing the weighted mean fluorescence lifetimes of all analyzed animals on day 4 or day 8 of life, respectively. Please click here to view a larger version of this figure.
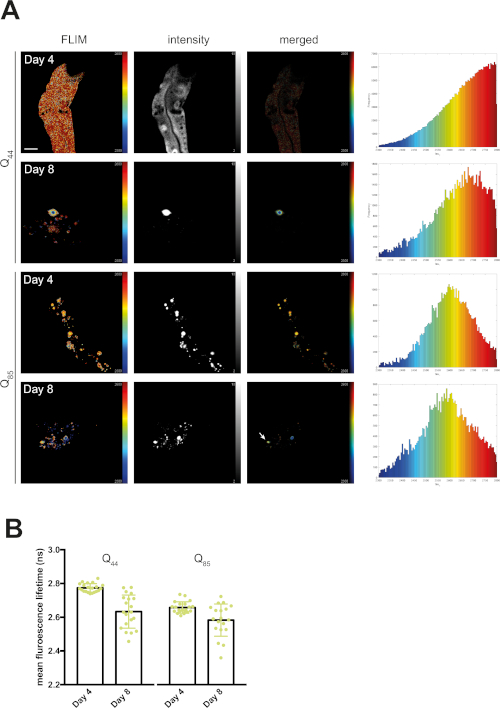
Figure 3: Fluorescence lifetimes of intestinal Q44-YFP and intestinal Q85-YFP decreased with age. (A) Representative maps of C. elegans expressing intestinal Q44-YFP or intestinal Q85-YFP on day 4 or day 8 of life generated by FLIMfit. Fluorescence lifetimes, fluorescence intensity, and a merged image of both are provided. Scale bars = 25 µm. Histograms show a distribution of measured lifetimes for all analyzed nematodes divided into 100 categories. (B) Bar plots showing the weighted mean fluorescence lifetimes of all analyzed animals on day 4 or day 8 of life, respectively. Please click here to view a larger version of this figure.
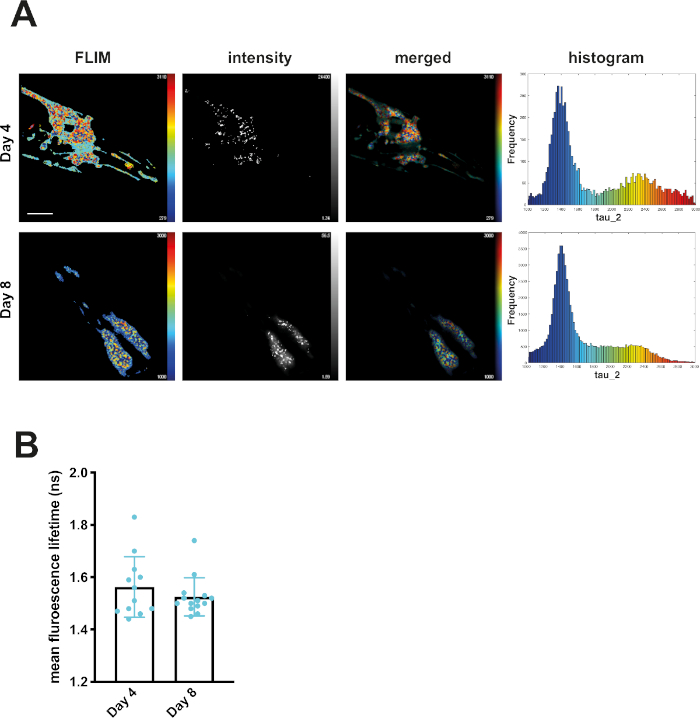
Figure 4: Fluorescence lifetimes of neuronal Q40-CFP did not change with age. (A) Representative maps of C. elegans expressing neuronal Q40-CFP on day 4 or day 8 of life generated by FLIMfit. Fluorescence lifetimes, fluorescence intensity, and a merged image of both are provided (the second lifetime, τ2, is indicated in all samples). Scale bars = 25 µm. Histograms show a distribution of measured lifetimes for all analyzed nematodes divided into 100 categories. (B) Bar plots showing the weighted mean fluorescence lifetimes of all analyzed animals on day 4 or day 8 of life, respectively. Please click here to view a larger version of this figure.
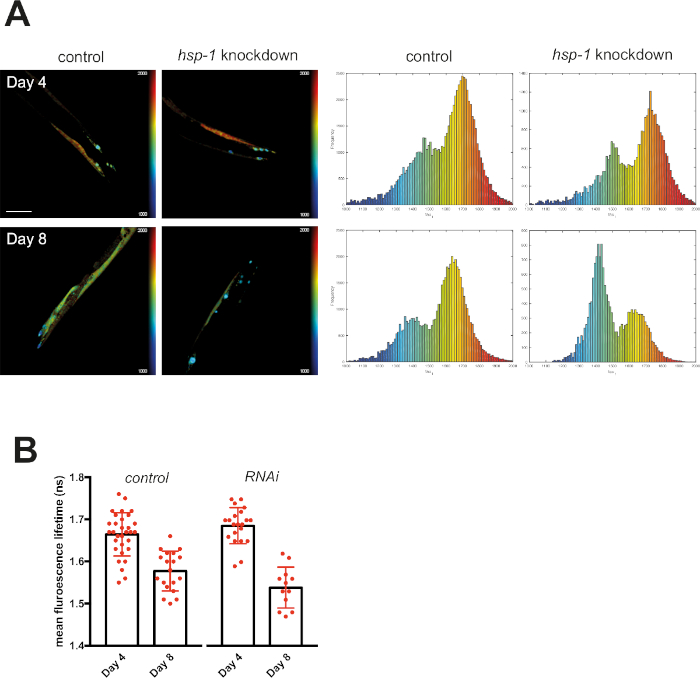
Figure 5: Fluorescence lifetimes of muscular Q40-RFP decreased upon knockdown of hsp-1. (A) Representative maps of C. elegans expressing muscular Q40-RFP on day 4 or day 8 of life generated by FLIMfit. A merge of the fluorescence lifetime and intensity map is displayed. For both time points, nematodes that grew on bacteria expressing an empty vector (control) or bacteria expressing the hsp-1 RNAi construct are shown. Scale bars = 25 µm. Histograms show a distribution of measured lifetimes for all analyzed nematodes divided into 100 categories. (B) Bar plots showing the weighted mean fluorescence lifetimes of all analyzed animals on day 4 or day 8 of life, with control or hsp-1 RNAi, respectively. Please click here to view a larger version of this figure.
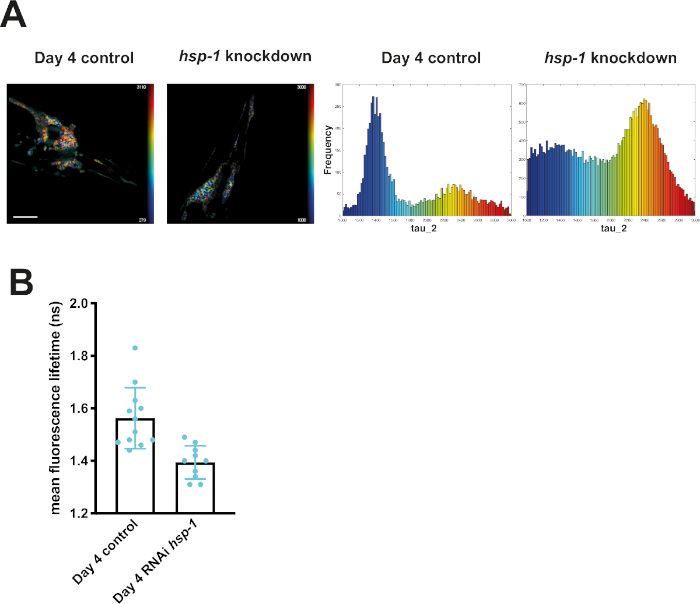
Figure 6: Fluorescence lifetimes of neuronal Q40-CFP decreased upon knockdown of hsp-1. (A) Representative maps of C. elegans expressing neuronal Q40-CFP on day 4 of life generated by FLIMfit. A merge of the fluorescence lifetime and intensity map is displayed. Nematodes shown were grown on bacteria expressing an empty vector (control) or bacteria expressing the hsp-1 RNAi construct. Scale bars = 25 µm. Histograms show a distribution of measured lifetimes for all analyzed nematodes divided into 100 categories. (B) Bar plots showing the weighted mean fluorescence lifetimes of all analyzed animals on day 4, with control RNAi or the hsp-1 RNAi. Please click here to view a larger version of this figure.
