Our automated cell culture and imaging system was designed to minimize human intervention allowing us to standardize the cultivation of hiPSC and differentiation into different cell types such as cortical or midbrain dopaminergic (mDA) neurons. A schematic overview of our automated cell culture system with integrated imaging devices is depicted in Figure 1. The initial introduction of cell cultures to this automated cell culture system can either be done by automatically seeding cells from a 50 mL tube or by using the “Loading Of Culture Plates” or “Loading Of Assay Plates” method for import of culture or assay plates. A central component of our system is the liquid handling station where all liquid transfer steps such as media changes or subcultivations are carried out. The custom-made deck layout of the liquid handler is represented in Figure 2. The liquid handling station is equipped with four positions. Up to four plates can be transferred from the incubator to the deck, allowing parallel media changes. Since in the subcultivation method, both the parent and daughter culture plates have to be accommodated on the deck, the maximum number of culture plates processed in parallel is limited to two. An important feature of the liquid handling station is the possibility to tilt the plates during media change for complete removal of cell culture supernatant. Also, the liquid handling station is equipped with shakers for favoring enzymatic dissociation of cells during the execution of the subcultivation protocol. Our automated culture system is also equipped with two imaging systems: a brightfield imaging cytometer for performing cell counting and confluency checks and, therefore, monitoring the cell growth over time, and a dual spinning disk confocal microscope for rapid, high content and high-resolution imaging of cells.
The hiPSC cultures are monitored daily for growth at the brightfield imaging cytometer and analyzed for percentage of confluency. The brightfield image in the left panel and in the right panel a green mask from the analysis of the brightfield image obtained with the cytometer (Figure 3A). A homogeneous hiPSC growth is observed over the time, as shown by the confluence percentages of two hiPSC lines (n = 4 plates) grown in parallel and subjected to confluency checks from day 1 to day 6 (Figure 3B). Upon reaching the set threshold, the hiPSC are passaged. The cell lines were cultured manually (m) or by the automation (a) system and observed for maintenance of typical stem cell morphology for at least two passages, representative brightfield images (Figure 4A). The hiPSC cultured manually (not shown) or in the automated system exhibited the typical stem cell marker OCT4 (red) and SSEA4 (green), as shown in the immunofluorescence assay (Figure 4B). The expression of the pluripotency markers OCT4, NANOG and REX1 were also assessed at mRNA level by qRT-PCR (Figure 4C). Relative quantifications were performed with samples collected from one cell line grown manually (m) and in the automated culture system (a) in duplicates (replicates 1 and 2). The expression levels of all three pluripotency markers in the replicates cultivated in the automated culture system are similar to marker expression after manual culture. On day 8 (D8), the expression of pluripotency markers was absent in cortical neurons differentiated (Diff) from hiPSC.
One important application of the automated culture system is the differentiation of hiPSC into different cell types including neurons. Here we show the differentiation of hiPSC into neurons using the NGN2 strategy, which produces a pure cortical neuron culture in a very short time (approximately 6 days). Neurons differentiated in the automated culture system (a) presented similar morphology and neuronal network organization as the neurons cultivated manually (m) (Figure 5A). Automated differentiated cortical neurons were positive for TUBB3 (neuron-specific Class III β-tubulin, red) and BRN2 (upper cortical layer marker, green) (Figure 5B), comparable to manually differentiated neurons (data not shown). The expression of neuronal markers including the microtubule-associated protein 2 (MAP2), the neural cell adhesion molecule (NCAM1) and Synapsin-1 (SYN1), as well as the cortical neuron markers BRN2 and CUX1 (upper cortical layer) were enriched in neurons at day 8 (D8) of differentiation (Figure 5C). Very low or no expression of these markers was observed in hiPSC. Relative quantifications were performed with samples collected from one cell line grown manually (m) and in the automated culture system (a) in duplicates (replicates 1 and 2). The expression levels in replicates show similar variations between manually and automated differentiations.
The integrated imaging capability of the automated culture system allows hands-free data collection for the health of cultures thus enabling long-term automated acquisition of phenotypic readouts. Using the NGN2 approach to a small molecule derived neural precursor (smNPC) line transduced with GFP lentivirus, we established a live-cell automated neurite outgrowth assay in which neurite length was measured over 11 days of differentiation without any manual intervention. The neurite complexity increased over time, as demonstrated by the area occupied with neurites on day 1, 3 and 11, GFP expression and masked images from analysis (Figure 6A, B). The increase in neurite length from day 1 to 11 of differentiation was quantified and showed a similar development across different wells. For the sake of simplicity, data from only 3 columns with 6 wells each from a 96-well plate is depicted in the representative graph although all inner 60 wells were analyzed (Figure 6C).
Another application of the automated culture system shown here is the differentiation of hiPSC into mDA neurons. The differentiation is based on media changes following a pre-established protocol and was performed on the automated culture system from days 0 to 65. Automated media changes did not cause cell detachment or any other visually detectable changes in the differentiation. At the end of the differentiation, on day 65, mDA neurons show cellular organization and morphology (spheric soma, long and spiny dendrites) comparable to manual differentiation (Figure 7A, B). At the mRNA level, mDA neurons differentiated in the automated culture system show the expression of neuronal and mDA markers, MAP2 and TH (tyrosine hydroxylase), respectively (Figure 7C). Both differentiations generated substantial amounts of TH and MAP2 positive neurons (Figure 7D).
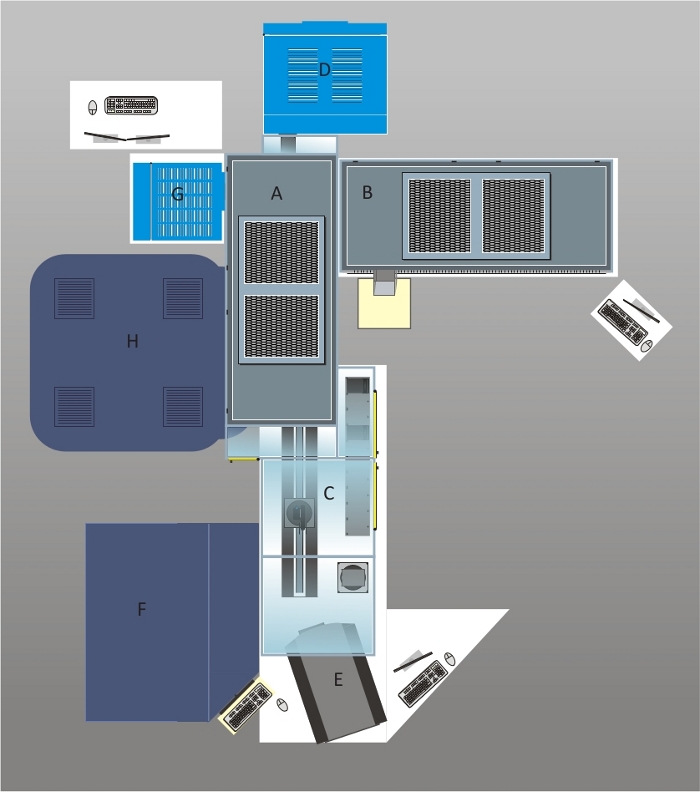
Figure 1: Schematic overview of the automated cell culture and imaging platform.
The system was designed with a polycarbonate housing and two HEPA hoods (A and B) equipped with four UV lamps ensuring a sterile environment for cell culture applications. Cell culture plates are loaded on shelves in front of the robotic arm which can be accessed via the front door (C). The plates are loaded into the CO2 incubator (D) with a capacity of 456 plates. A brightfield cell cytometer (E) is used for confluency checks and cell counting during subcultivation routines. The liquid handling station is below one of the HEPA hoods (B). The deck layout of the liquid handler is described in Figure 2. The pipetting arm of the liquid handling station carries a 96 channel pipetting head, eight 1 mL pipetting channels and four 5 mL pipetting channels. In the case of the 1 mL pipetting channels, tips or needles can be used for liquid transfers. For screening purposes, cells seeded in assay plates can be treated with samples stored at -20 °C in the automated -20 °C storage system (F) after thawing these samples in a second incubator (G). High throughput imaging is performed in the automated confocal microscope (H) offering to acquire images in confocal mode using two spinning discs or in epifluorescence mode. A live-cell chamber integrated into the microscope allows performing long-term imaging of cultured cells. Please click here to view a larger version of this figure.
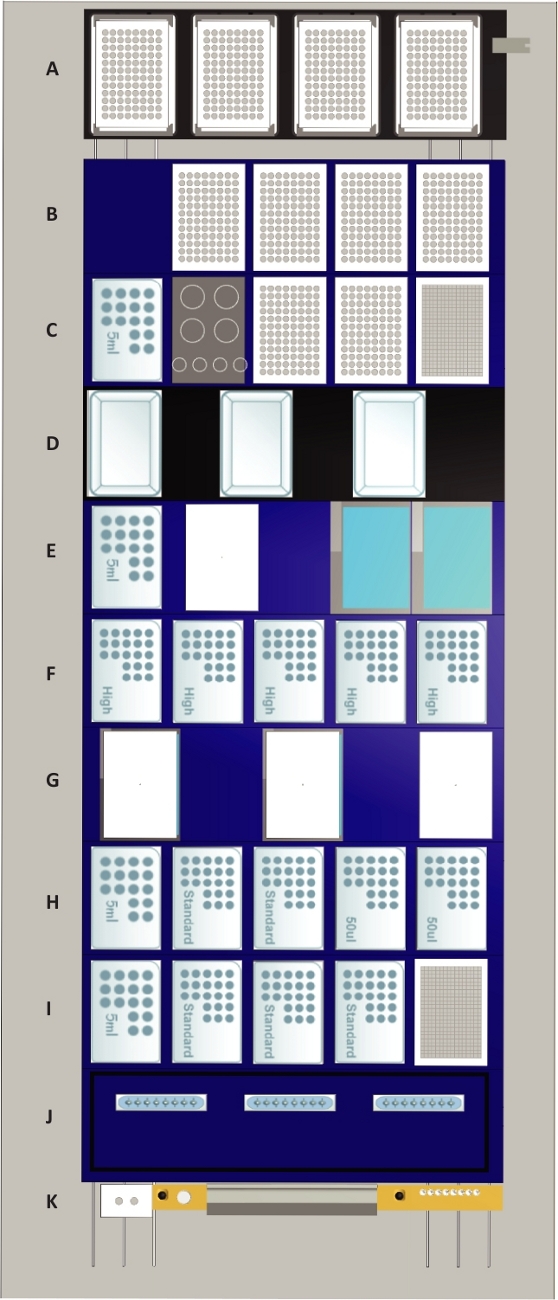
Figure 2: The deck layout of the liquid handling station.
Tip positions are indicated by “50 µL” for 50 µL tips, by “standard” for 300 µL tips, by “high” for 1 mL tips and by “5 mL” for 5 mL tips. Deck components: (A) Four heated shaker positions (max speed: 2500 rpm) which can be used for any culture plate or assay plate format. The shaker positions are equipped with clampable grippers which are also used for plate alignment following transports to the deck. Furthermore, the shaker positions function as a lid parking position for plates during liquid transfer steps. For representative purposes, all shaker positions are occupied by 96 well assay plates. (B) Four tilt modules for processing four plates of any format simultaneously are positioned on the top. The lowest position marks the waste collection chamber for culture and assay plates and the 96 channel pipetting head. For representative purposes, all tilt modules are occupied by 96-well assay plates. (C) On the top position, the 384-well plate for cell counting is located. Below are two positions for plates that are occupied by 96-well plates for representative purposes and a rack for four 50 mL and four 15 mL tubes. The lowest position is occupied by 5 mL tips. (D) Three media lines with positions for media reservoirs. The media lines possess liquid level sensors that allow to automatically fill the media reservoir with up to 250 mL of media. (E) Two liquid waste modules with active drain are based on the top, below a temperature-controlled module with a position for one container (white) and a 5 mL tip rack are located. (F) Five positions for 1 mL tips. (G) Two temperature-controlled modules are positioned at the bottom and a position for parking one of their lids on the top. (H) Positions for two 50 µL nested tip racks (NTR) in the top followed by two positions for single-channel and 96-channel pick up of 300 µL tips and a 5 mL tip rack at the bottom. (I) A stacker for 384-well counting plates at the top followed by three 300 µL NTR and a 5 mL tip rack at the bottom. (J) Storage and wash station for three sets of eight reusable metal 1 mL needles. (K) Waste position for 1 mL and 5 mL pipetting channels and empty NTR (grey) as well as a gripper block for 1 and 5 mL channels (white) used for on-deck transport steps. Please click here to view a larger version of this figure.
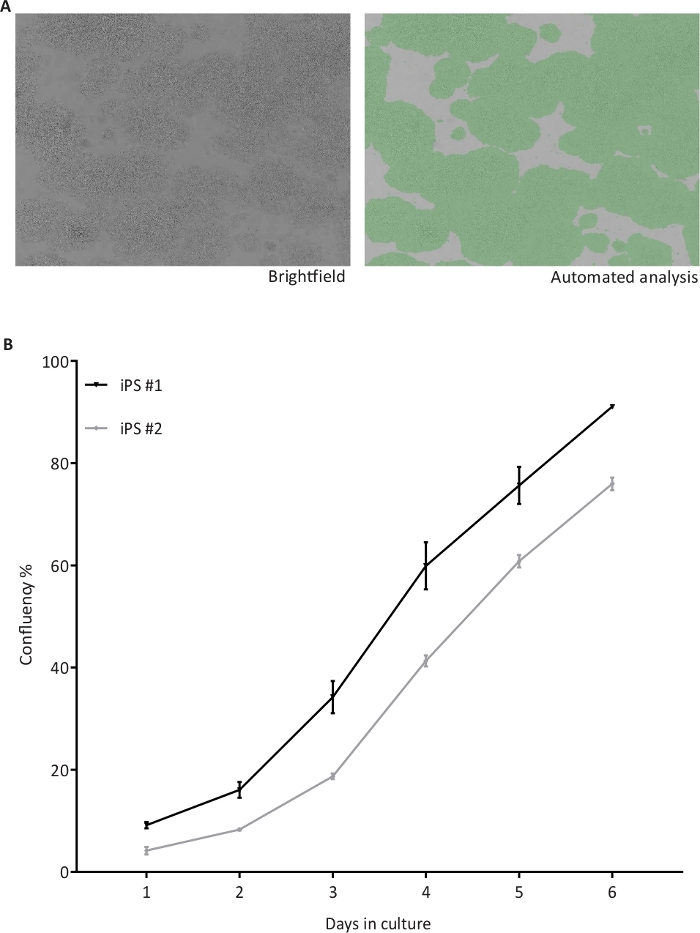
Figure 3: Automated confluency check of hiPSC.
(A) Representative brightfield (BF) images of hiPSC taken by the cell cytometer (left) and after automated confluency analysis (right) indicating the proportional area occupied by cells in green; (B) Confluency percentages recorded from two hiPSC lines (iPS #1 and #2) from day 1 to 6 of culture, n = 4 1-well plates per cell line. Please click here to view a larger version of this figure.
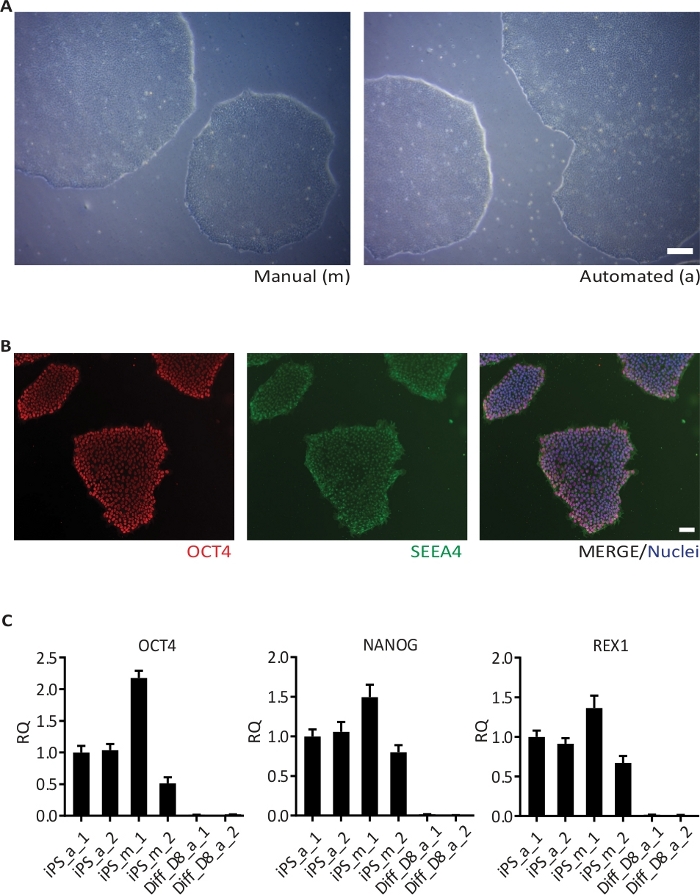
Figure 4: Passaging of hiPSC.
(A) BF image of one hiPSC line grown manually (left) and using the automated culture system (right). Images were taken 6 days after the second passaging; (B) Representative images of hiPSC stained for the pluripotency markers OCT4 and SSEA4, and counterstained with Hoechst 33342 (Nuclei); (C) Results of qRT-PCR for the pluripotency markers OCT4, NANOG and REX1 in one hiPSC line cultivated in duplicates (1 and 2) manually (m) and in the automated culture system (a), and the respective hiPSC-derived cortical neurons (Diff) at day 8 (D8) of differentiation. The data is represented as the relative quantity (RQ) using iPS_a_1 as the reference sample. Error bars represent standard deviation (SD) from 3 technical replicates of qRT-PCR reaction. GAPDH, RPL13A1 and RPLPO were used as housekeeping genes. Scale bar: 100 µm. Please click here to view a larger version of this figure.
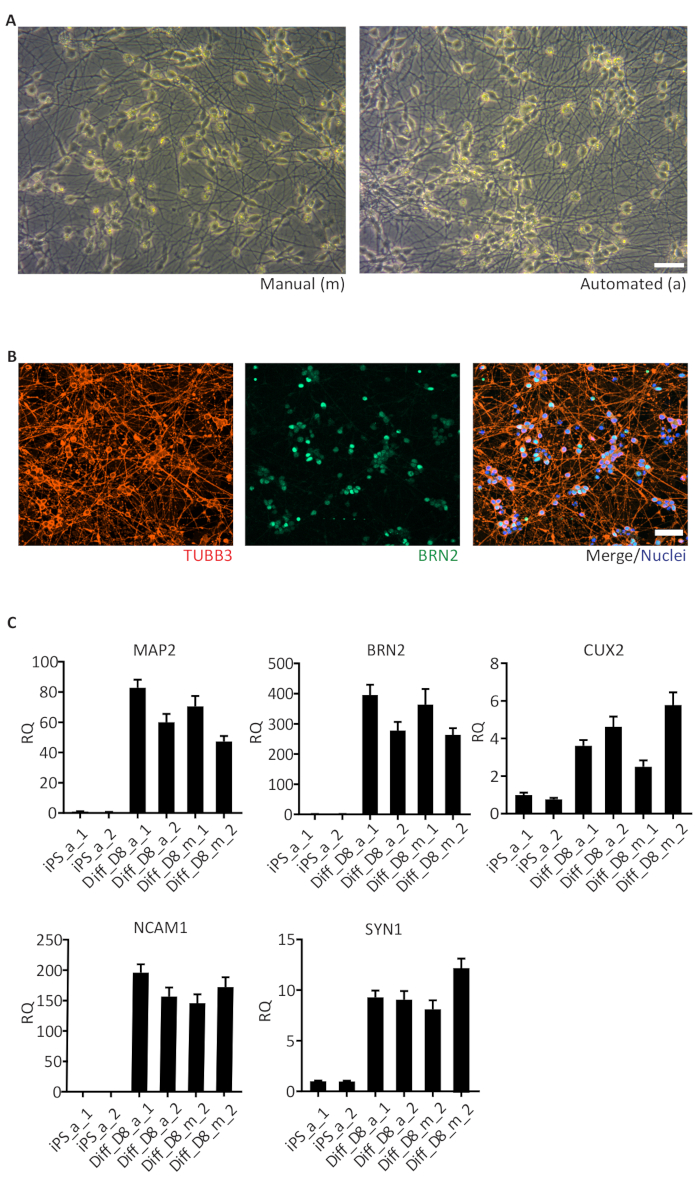
Figure 5: Human iPSC-derived cortical neurons.
(A) Brightfield images of day 6, manually (m) and automated (a) differentiated cortical neurons showing similar neuronal networks; (B) Representative images of cells stained for TUBB3 (pan neuronal), BRN2 (cortical neurons) and Hoechst 33342 (nuclei) on day 8 of differentiation; (C) Results of qRT-PCR for marker genes of cortical neurons (MAP2, BRN2, CUX2, NCAM1 and SYN1) enriched in day 8 (D8) of differentiation. The data is represented as the relative quantity (RQ) using iPS_a_1 as the reference sample. Error bars represent the SD from 3 technical replicates of qRT-PCR reaction. GAPDH, RPL13A1 and RPLPO were used as housekeeping genes. Scale bar: 50 µm. Please click here to view a larger version of this figure.
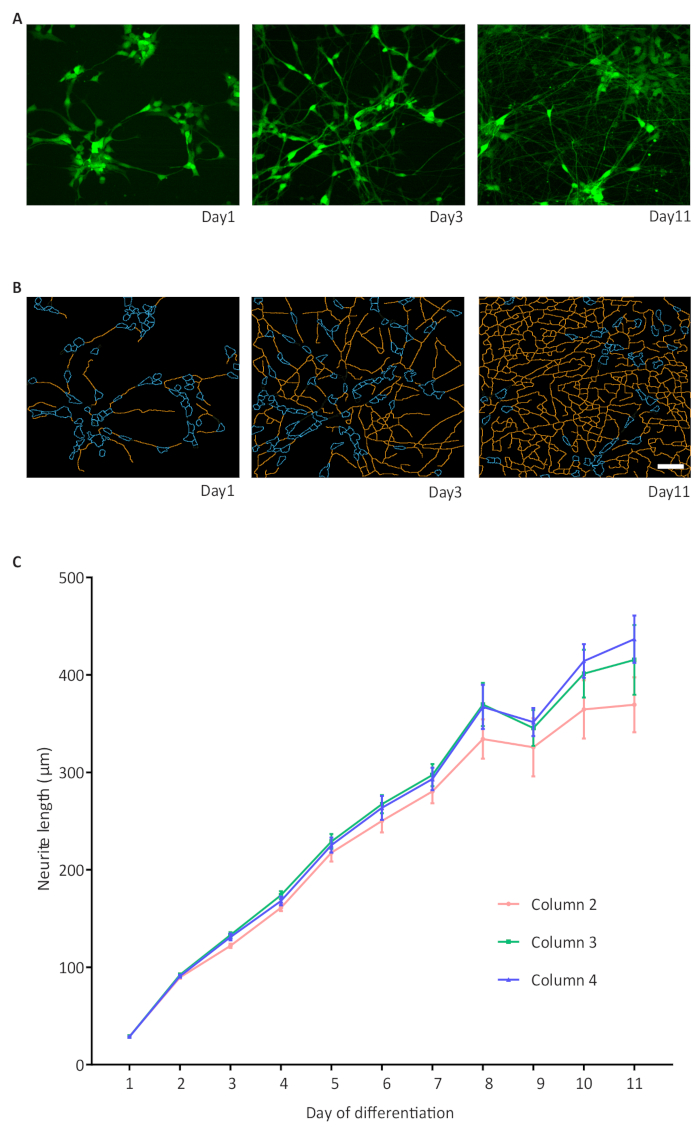
Figure 6: A high-throughput assay for neurite outgrowth.
(A) Representative images of GFP expressing cells on days 1, 3 and 11 of differentiation; (B) Representative binary images of neurites on days 1, 3 and 11 of differentiation. The neurite outgrowth was quantified using the high content image analysis software 1 and is represented as neurite length; (B) The graphic displays the increase in neurite length in NPC-derived NGN2 neurons and formation of a dense network. Three 96-well plates with cells in the inner 60 wells are imaged. For simplicity, only three columns of wells per 96-well plate are shown as an example with n = 6 wells per column. An average of 1308 cells were analyzed per well. Error bars represent the standard error of the mean (S.E.M.). Scale bar = 50 µm. Please click here to view a larger version of this figure.
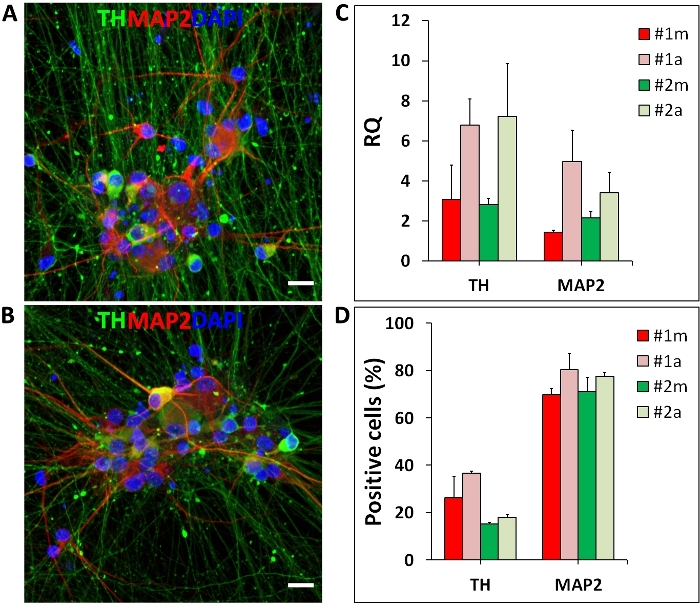
Figure 7: Human iPSC-derived mDA neurons.
Midbrain DA neurons differentiated manually (m) and in the automated (a) culture system. (A, B) Representative fluorescent images of mDA neurons stained for tyrosine hydroxylase (TH, mDA neuron marker; green), MAP2 (neuronal marker; red) and Hoechst 33342 (nuclei; blue); (C) Representative qRT-PCR results for marker genes of mDA neurons differentiated manually and in the automated culture system. TH and MAP2 expression levels are represented as relative quantity (RQ) normalized to housekeeping genes (OAZ1 and GAPDH); (D) Percentages of TH and MAP2 positive neurons generated by manual and automated differentiation. Error bars represent the SD of two independent differentiations performed with two distinct iPSC lines (#1 and #2). Scale bar = 50 µm. Please click here to view a larger version of this figure.
| Cell type | Purpose | Protocol step | Cell density | Plate format | Cell number/well |
| NGN2 differentiation | |||||
| iPSC | NGN2 stable line generation | S.2.3. | 30,000 cells/cm2 | 12-well | 1,17,000 |
| iPSC | NGN2 neuron differentiation | 3.1.3. | 30,000 cells/cm2 | 1-well | 25,20,000 |
| iPSC | NGN2 neuron differentiation | 3.1.3. | 30,000 cells/cm2 | 96-well | 9,600 |
| smNPC generation | |||||
| smNPC | Replating day 12 and 16 | S5.9. and S5.11. | 70,000 cells/cm2 | 6-well | 6,72,000 |
| smNPC | Replating from passage 5 | 5.11. | 50,000 cells/cm2 | 6-well | 4,80,000 |
| smNPC | smNPC to NGN2 neurons | 3.2.2 | 50,000 cells/cm2 | 96-well | 16,000 |
| mDA differentiation | |||||
| iPSC | mDA neuron differentiation | 4.1.2. | 200,000 cells/cm2 | 1-well | 1,68,00,000 |
| DA neurons | Day 25 replating | 4.3.2. | 400,000 cells/cm2 | 1-well | 3,36,00,000 |
| DA neurons | Day 25 replating | 4.5.2. | 100,000 cells/cm2 | 96-well | 32,000 |
| Coating | Purpose | Protocol step | Concentration/ Dilution |
Plate format | Details of coating |
| iPS culture | |||||
| Extracellular matrix | iPSC, NGN2 line, mDA neurons |
1.5.7. and S2.3. | 1 aliquot*; 25 mL DMEM/F-12 |
1-/12-well | 8/0.5 mL/well; 1 h at RT |
| NGN2 differentiation | |||||
| Poly-L-Ornithine | NGN2 neuron differentiation | 3.1.3. and 3.2.2. | 0.1 mg/mL; PBS | 1-/96-well | 8/0.1 mL/well; 12 h at 37°C; 3x PBS wash |
| Laminin | NGN2 neuron differentiation | 3.1.3. and 3.2.2. | 5 μg/mL; PBS | 1-/96-well | 8/0.1 mL/well; 4 h at 37°C |
| smNPC generation | |||||
| Extracellular matrix | smNPC generation and culture | S5.6., S5.9., S5.11. |
1 aliquot*; 25 mL DMEM/F-12 |
6-well | 1 mL; 2 h at RT |
| mDA differentiation | |||||
| Extracellular matrix | mDA differentiation | 4.1.2. | 1 aliquot*; 25 mL DMEM/F-12 |
1-well | 12 mL; 12 h at 37°C |
| Poly-L-Ornithine | mDA differentiation | 4.3.2. and 4.5.2. | 0.1 mg/mL; PBS | 1-/96-well | 12/0.1 mL/well; 12 h at 37°C; 3x PBS wash |
| Laminin | mDA differentiation | 4.3.2. and 4.5.2. | 10 μg/mL; PBS | 1-/96-well | 12/0.1 mL/well; 12 h at 37°C |
| Fibronectin | mDA differentiation | 4.3.2. and 4.5.2. | 2 μg/mL; PBS | 1-/96-well | 12/0.1 mL/well; 12 h at 37°C |
| *A extracellular matrix aliquot is defined as the dilution factor (in µL) present in the Certificate of Analysis of this product. | |||||
Table 1: Seeding cell density and coating respective to the plate format.
| Day | Reagent | ||
| Day 0 – 1 | 100 nM LDN193189, 10 µM SB431542 | ||
| Day 1 – 3 | 100 nM LDN193189, 10 µM SB431542, 1 mM SHH, 2 mM Purmorphamine, 100ng/mL FGF-8b |
||
| Day 3 – 5 | 100 nM LDN193189, 10 µM SB431542, 1 mM SHH, 2 mM Purmorphamine, 100ng/mL FGF-8b, 3 µM CHIR99021 |
||
| Day 5 – 7 | 100 nM LDN193189, 1 mM SHH, 2 mM Purmorphamine, 100ng/mLFGF-8b, 3 µM CHIR99021 |
||
| Day 7 – 9 | 100 nM LDN193189, 1 mM SHH, 3 µM CHIR99021 | ||
| Day 9 – 11 | 100 nM LDN193189, 1 mM SHH, 3 µM CHIR99021 | ||
| Day 11 – 13 | 3 µM CHIR99021, 20 ng/mL BDNF, 0.2 mM L-ascorbic acid (AA1), 20 ng/mL GDNF, 1 mM db-cAMP, 1 ng/mL TGFß3, 10 µM DAPT |
||
| Day 13 – 65 | 20 ng/mL BDNF, 0.2 mM L-ascorbic acid (AA1), 20 ng/mL GDNF, 1 mM db-cAMP, 1 ng/mL TGFß3, 10 µM DAPT |
||
Table 2: Small molecule addition for dopaminergic neuron differentiation.
| Day | KSR medium | N2 medium | Differentiation medium |
| Day 0 – 1 | 100% | 0 | 0 |
| Day 1 – 3 | 100% | 0 | 0 |
| Day 3 – 5 | 100% | 0 | 0 |
| Day 5 – 7 | 75% | 25% | 0 |
| Day 7 – 9 | 50% | 50% | 0 |
| Day 9 – 11 | 25% | 75% | 0 |
| Day 11 – 13 | 0 | 0 | 100% |
| Day 13 – 65 | 0 | 0 | 100% |
Table 3: Media gradient for dopaminergic neuron differentiation.
Supplementary File 1. Please click here to download this file.
